Синдром врожденного косого диска зрительного нерва
Добавил пользователь Дмитрий К. Обновлено: 28.01.2026
Синдром врожденного косого диска зрительного нерва
Синдром косого диска — не наследственная двусторонняя аномалия, при которой верхневисочная часть диска проминирует, а нижненосовая часть смещена назад, в результате чего формируется диск зрительного нерва овальной формы, его длинная ось ориентирована косо; эти изменения сопровождаются situs inversus сосудов сетчатки, врожденным нижненосовым конусом, истончением пигментного эпителия сетчатки (ПЭС) и хориоидеи нижненосового квадранта и битемпоральной гемианопсией. Изменения диска зрительного нерва развиваются вторично вследствие задней эктазии нижненосовой части глазного дна и диска зрительного нерва (ДЗН).
Вследствие региональной эктазии глазного дна у пациентов отмечается миопический астигматизм, «плюсовая» ось ориентирована параллельно эктазии. Вклад в формирование астигматизма вносит неправильная кривизна роговицы, выявляемая при кератотопографии. Локализация экскавации в нижненосовом или нижнем квадранте указывает на связь этой аномалии с колобомой, характер этой связи не установлен.
У пациентов может наблюдаться битемпоральная гемианопсия или проминирование диска зрительного нерва, симулирующее застой диска зрительного нерва (ДЗН). Битемпоральная гемианопсия обычно неполная, и ограничена преимущественно верхними квадрантами, причиной ее является рефракционная скотома, возникающая вследствие региональной миопии в нижненосовой зоне сетчатки.
В отличие от поражений хиазмы, при кинетической периметрии граница этих дефектов поля зрения совпадает с вертикальным меридианом. Кроме того, снижение чувствительности в верхневисочном квадранте селективно ограничено средней изоптерой, тогда как чувствительность по большой и малой изоптерам остается совершенно нормальной, вследствие выраженной эктазии глазного дна на средней периферии. При повторной периметрии с линзой -4,0 аномалии полей зрения часто исчезают. В некоторых случаях в зоне эктазии чувствительность сетчатки снижена, и этот дефект не исчезает полностью даже после соответствующей оптической коррекции.
У нескольких пациентов синдром косого диска сопровождался истинной битемпоральной гемианопсией, позже у этих больных были диагностированы супраселлярные опухоли. Эти аномалии могли развиться вследствие нарушения миграции аксонов зрительного нерва под действием опухоли в процессе эмбриогенеза. Поэтому пациентам с синдромом косого диска зрительного нерва, у которых битемпоральная гемианопсия ограничена вертикальным меридианом или при кинетической периметрии не наблюдается дефекта преимущественно по среднепериферической изоптере, показано лучевое исследование. Косой диск без эктазии сетчатки наблюдается у пациентов с транссфеноидальным эцефалоцеле.
Синдром косого диска также был описан при Х-сцепленной врожденной стационарной ночной слепоте. Аномалии на границе стафиломы или между перипапиллярной сетчаткой и измененными краями диска могут стать причиной серозной отслойки макулярной зоны сетчатки.
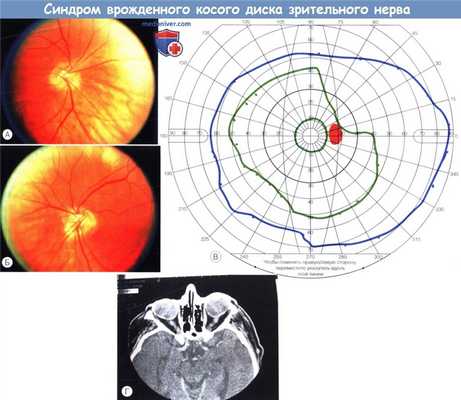
Синдром врожденного косого диска.
(А) Правый диск зрительного нерва.
(Б) Левый диск зрительного нерва.
(В) Поле зрения правого глаза, виден верхневисочный дефект, ограниченный среднепериферической изоптерой, не пересекающий вертикальный меридиан.
(Г) На компьютерной томограмме видно увеличение кривизны заднего отдела склеры с носовой стороны и уплощение задней части склеры с височной стороны.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Aicardi у ребенка
Синдром Aicardi — это заболевание головного мозга и сетчатки неизвестной этиологии. Клинические проявления: инфантильные спазмы, агенезия мозолистого тела, электроэнцефалографический паттерн, называющийся гипсаритмией, и множественные депигментированные «хориоретинальные лакуны» вокруг диска зрительного нерва.
Гистологически хориоретинальные лакуны представляют собой четко отграниченные сквозные дефекты пигментного эпителия сетчатки и сосудистой оболочки. Покрывающая лакуны сетчатка остается интактной, но при гистологическом исследовании часто выявляются ее аномалии.
Хориоретинальные лакуны могут сочетаться с гипоплазией зрительного нерва, колобомой диска зртельного нерва (ДЗН) и врожденной пигментацией диска зрительного нерва. Другие аномалии глаз включают в себя микрофтальм, ретробульбарную кисту, псевдоглиому, отслойку сетчатки, рубцы макулярной области, катаракту, зрачковые мембраны, колобомы и синехии радужки.
Наиболее часто встречающиеся системные аномалии — это мальформации позвоночника (например сросшиеся позвонки, сколиоз, незаращение дужек позвонков) и ребер (например отсутствие ребра, сросшиеся или раздвоенные ребра), мышечная гипотония, микроцефалия, дисморфия лица и аномалии наружного уха. Почти всегда наблюдается тяжелая умственная отсталость. У пяти пациентов описана папиллома сосудистого сплетения желудочков мозга.
Аномалии ЦНС при синдроме Aicardi включают в себя агенезию мозолистого тела, аномалии миграции клеток коры (например пахигирию, полимикрогирию, кортикальные гетеротопии) и множественные структурные мальформации ЦНС (например асимметрию полушарий головного мозга, вариант Dandy-Walker, кольпоцефалию, срединные кисты паутинной оболочки). Выявлено частичное совпадение синдрома Aicardi и септо-оптической дисплазии.
Причиной синдрома Aicardi может быть Х-сцепленная мутация, приводящая к гибели мальчиков. Необходимо расспросить родителей о наличии в анамнезе невыношенных беременностей. Скорее всего, все случаи синдрома Aicardi развиваются в результате новых мутаций, поскольку случаи развития заболевания у сиблингов встречаются редко. Синдром Aicardi у двух сестер указывает на наличие у родителя гонадного мозаицизма по патогенной мутации.
Хотя ранние инфекции ЦНС могут вызывать тяжелые аномалии, результаты тестов на возбудителей инфекций стабильно отрицательны. Не выявлено связи синдрома Aicardi с воздействием тератогенных препаратов или других токсических веществ. Характер мальформаций головного мозга и сетчатки указывает на поражение ЦНС в сроки между четвертой и восьмой неделями гестации.
У большинства детей развиваются некупируемые припадки, 91% пациентов достигают вех развития, соответствующих не более чем годовалому возрасту. Внутриглазные аномалии включают в себя микрофтальм, персистирующую зрачковую мембрану, персистирующее гиперпластическое первичное стекловидное тело, сосудистые петли диска зрительного нерва и эпиретинальную глиальную ткань.
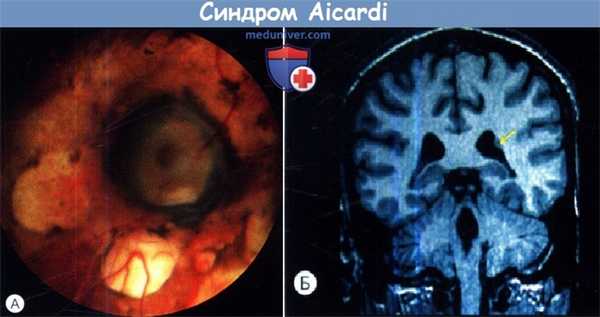
Синдром Aicardi.
(А) Перипапиллярные хориоретинальные лакуны вокруг диска зрительного нерва.
(Б) На МРТ видна агенезия мозолистого тела.
Гипоплазия зрительного нерва
Гипоплазия зрительного нерва – врожденная аномалия, характеризующаяся уменьшением количества аксонов в структуре пораженного нерва. Клиническая симптоматика представлена снижением остроты зрения, нистагмом, косоглазием, повышенной утомляемостью. Диагностика основывается на проведении прямой офтальмоскопии, электроретинографии, КТ головы, визометрии, гистологического и микроскопического исследования. Пациентам показана электростимуляция зрительного нерва, лазерная плеоптика, компенсация депривации, а также симптоматическая терапия страбизма и нистагма.

Общие сведения
Гипоплазия зрительного нерва впервые была описана британским офтальмологом В. Ньюменом в 1864 году. Заболевание диагностируют у 50% детей с диагнозом фетального алкогольного синдрома. Согласно статистическим данным, в возрасте от 5 до 15 лет 5,9% от всех случаев слепоты обусловлены этой аномалией развития глаза. Около 90% детей с этой патологией страдают страбизмом или нистагмом. У 46-53% больных помимо недоразвития оптических волокон диагностируют агенезию мозолистого тела и прозрачной перегородки. У 12-45% пациентов удается выявить ряд мальформаций центральной нервной системы. Заболевание с одинаковой частотой встречается среди лиц мужского и женского пола.
Причины гипоплазии зрительного нерва
Иногда при недоразвитии оптических волокон обнаруживается мутация гена РАХ6 в локусе 11р13, которая обычно сочетается с помутнением хрусталика и аниридией. Установлен аутосомно-доминантный тип наследования. К пусковым факторам относятся:
- Воздействие тератогенных факторов. Экспериментальным путем было доказано, что прием спиртных напитков, потребление наркотических средств (кокаин) и курение в период беременности достоверно повышает вероятность возникновения заболевания.
- Метаболические расстройства. Патологию часто диагностируют у детей, рожденных от матерей с сахарным диабетом 1 типа, гипотиреозом в анамнезе.
- Влияние лекарственных средств. Поражение оптических нервных волокон у плода вызывает прием матерью фенобарбитала, антидепрессантов, хинина во время беременности.
- Внутриутробные инфекции. Развитие болезни часто провоцирует инфицирование плода цитомегаловирусом, герпетической инфекцией.
Патогенез
Патогенетический механизм напрямую связан с нарушением деления ганглиозных клеток внутренней оболочки глаза на 4-6 неделе эмбриогенеза. Амакриновые и горизонтальные клетки не поражаются. Согласно другой патогенетической теории, заболевание вызвано феноменом аксональной регрессии на 16-31 гестационной неделе. При сочетании болезни с другими аномалиями развития головного мозга ведущая роль в формировании дефектов отводится ретроградной дегенерации, обусловленной преобладанием энцефалокластических процессов. Нарушение дифференциации структур полушарий и оптических волокон зачастую связано с патологией регуляторных процессов. Механическая теория базируется на компрессии зрительного пути злокачественным новообразованием, организовавшимся экссудатом, сгустками крови.
Симптомы гипоплазии зрительного нерва
В случае тяжелого течения первые клинические проявления родители наблюдают уже в период новорожденности. У детей рано развивается косоглазие, нистагм, нарушение ориентации в пространстве. В старшем возрасте пациенты предъявляют жалобы на двоение перед глазами, головокружение, затруднения при фиксации взгляда, которые проявляются мелкоразмашистыми движениями. Симптоматика усиливается при волнении, усталости, в стрессовых ситуациях. При длительной концентрации взгляда возникает головная боль, затуманивание зрения, повышенная утомляемость. Нарушение остроты зрения варьирует от незначительного снижения до полной слепоты.
Патология может иметь одно- или двухсторонний характер. При односторонней или асимметричной форме нарушается бинокулярное зрение. При закрытии больного глаза симптомы исчезают. Часто дети пытаются ограничить участие пораженного глазного яблока в акте зрения. С целью компенсировать зрительную дисфункцию пациенты наклоняют голову под углом наиболее четкого видения. Для двухсторонней формы характерна девиация глаза, которая проявляется постоянным отклонением глазных яблок в определенную сторону. При этом создается впечатление, что человек всегда смотрит в одном направлении. У большинства пациентов выявляется афферентный зрачковый дефект. Из-за несимметричного положения радужной оболочки и зрачка часто наблюдаются затруднения адаптации в социуме.
Осложнения
Гипоплазия зрительного нерва часто осложняется выраженным снижением зрения вплоть по амавроза. Пациенты с данной патологией в анамнезе более склонны к присоединению инфекционных и воспалительных заболеваний глаз (пан- и эндофтальм, склерит), что связано с нарушением регионального кровоснабжения и трофики. Как правило, вторичное вовлечение в патологический процесс сетчатой оболочки и увеального тракта ведет к развитию ретинита, переднего и заднего увеита. При сопутствующем поражении оптических трактов возникает гемианопсия. Заболевание нередко сочетается со злокачественными новообразованиями головного мозга (супраселлярные опухоли, тератома).
Диагностика
Для постановки диагноза применяют физикальный осмотр и специальное офтальмологическое обследование. При наличии патологии у родителей или близких родственников на 16 неделе внутриутробного развития осуществляют генетический скрининг. В постнатальном периоде диагностика базируется на проведении:
Дополнительно показан расчет отношения расстояния между ДЗН и макулой к диаметру диска, который в норме составляет менее 3. Дифференциальная диагностика проводится с аплазией и атрофией оптического нерва. Характерные офтальмоскопические признаки атрофии – восковый оттенок ДЗН, экскавация его поверхности, сужение и уменьшение числа сосудов сетчатки. При аплазии определяется полное отсутствие диска зрительного нерва на фоне слабо выраженной пигментации внутренней оболочки глазного яблока. Макула не подлежит дифференциации. Визуализируются только сосуды хориоидеи.
Лечение гипоплазии зрительного нерва
Терапия результативна только при раннем проведении лечебных мероприятий. Это связано с тем, что в на первом году жизни пре- и постгеникулярные пути, латеральное коленчатое тело и корковые центры находятся на стадии формирования. Лечение включает в себя:
- Чрескожную электростимуляцию зрительного нерва. Эффективность метода напрямую зависит от исходной остроты зрения. Электростимуляция назначается курсами. В среднем проводится от 3 до 5 курсов.
- Компенсацию глазной депривации. Для компенсации этого явления ребёнку проводят очковую и контактную коррекцию остроты зрения. Профилактика развития амблиопии предполагает использование дозированной окклюзии лучше видящего глаза.
- Лазерную плеоптику. Методика основывается на применении лазерного излучения низкой интенсивности, которое позволяет улучшить микроциркуляцию и метаболические процессы в окружающих тканях, повышает активность ДНК, РНК и ферментов, благотворно влияет на энергетический потенциал клеток и трофические процессы.
Симптоматическая терапия базируется на хирургическом лечении нистагма и страбизма. Оперативные способы коррекции косоглазия используются в возрасте до 5 лет. В зависимости от типа страбизма назначают операции, которые ослабляют или усиливают функции глазодвигательных мышц. Цель лечения при нистагме – формирование позиции относительного покоя путем восстановления физиологического положения мышц. По индивидуальным показаниям осуществляют инъекции ботокса в глазничную полость для уменьшения амплитуды мелкоразмашистых движений глазных яблок. Дополнительно проводят коррекцию нейроэндокринных нарушений (неонатальная гипогликемия, пангипопитуитаризм, вторичный гипотиреоз).
Прогноз и профилактика
Прогноз определяется степенью выраженности гипоплазии. Незначительный дефект строения зрительного нерва может долгое время оставаться незамеченным. Своевременно начатое лечение приводит к положительным результатам. Специфическая профилактика этой аномалии развития не разработана. Неспецифические превентивные меры сводятся к предупреждению воздействия тератогенных факторов (спиртные напитки, наркотические средства, ионизирующее излучение). При высоком риске возникновения болезни показано проведение перинатальной диагностики и специфической терапии с периода новорожденности.
Септооптическая дисплазия
Септооптическая дисплазия – врожденное заболевание, относящееся к порокам прозэнцефалической группы, характеризуется аномалиями развития зрительного нерва, гипофиза и прозрачной перегородки. Симптомами этого состояния являются нистагм и другие зрительные нарушения, признаки эндокринных расстройств (задержка роста и полового созревания), возможно развитие умственной отсталости. Диагностика септооптической дисплазии производится на основании данных общего осмотра больного, офтальмологических исследований, компьютерной и магнитно-резонансной томографии головного мозга, а также молекулярно-генетических анализов. Специфическое лечение не разработано, симптоматические мероприятия включают в себя коррекцию зрения и заместительную гормональную терапию при эндокринных расстройствах.
Септооптическая дисплазия (синдром де Морсье) – врожденное нарушение развития головного мозга и зрительного аппарата различной (в том числе и генетической) природы. Причинами этого состояния, помимо генетических мутаций, могут выступать инфекции матери во время вынашивания ребенка, молодой возраст родителей, сосудистые нарушения у женщины. Название «септооптическая дисплазия» ввел французский педиатр Де Морсье, который в 1956 году составил наиболее обширное и полное описание этого состояния. В настоящий момент под синдромом Де Морсье врачи-генетики подразумевают только наследственную форму септооптической дисплазии. На сегодняшний день общая встречаемость этого заболевания (как приобретенных, так и наследственных форм) составляет примерно 1 случай на 10 000 новорожденных, мальчики и девочки поражаются с одинаковой частотой. Выраженность септооптической дисплазии может значительно отличаться у разных больных – от практически полного отсутствия симптомов до тяжелых нарушений, осложненных ДЦП, отставанием в физическом и умственном развитии.
Причины септооптической дисплазии
Септооптическая дисплазия наследственного характера развивается по причине мутаций в гене HESX1, расположенного на 3-й хромосоме. Этот ген принадлежит к обширному классу гомеобоксных генов, принимающих активное участие в регуляции процессов эмбриогенеза. В частности, HESX1 регулирует эмбриональное развитие структур головного мозга (прозрачной перегородки, гипофиза, хиазмы, зрительного нерва), поэтому его мутации приводят к развитию септооптической дисплазии. Кроме этого, доказано участие данного гена в регуляции осевой и двухсторонне-симметричной структуры тела. На сегодняшний день выявлено четыре типа миссенс-мутаций гена HESX1, наличие которых обуславливает появление признаков септооптической дисплазии, все они наследуются по аутосомно-рецессивному типу.
Выраженность симптомов заболевания может существенно различаться – от незначительных зрительных нарушений (миопии, косоглазия) до яркой клинической картины со слепотой, гипопитуитаризмом и тяжелой умственной отсталостью. Исследователи пока не нашли взаимосвязи между типом мутации HESX1 и тяжестью симптомов септооптической дисплазии. Возможно, развитие этого заболевания является результатом совокупного влияния как внутренних (генетических), так и внешних факторов. Также остается неясным то обстоятельство, что у детей молодых матерей (в возрасте менее 23 лет) септооптическая дисплазия возникает чаще, а ее наследственная разновидность протекает намного тяжелее.
Патогенез септооптической дисплазии заключается в нарушении процесса дифференцировки эмбриональных тканей в области зачатков гипофиза, хиазмы, мозолистого тела и других структур мозга. По этой причине данное заболевание в тяжелых случаях может сопровождаться другими неврологическими симптомами – детским церебральным параличом, умственной отсталостью. Практически всегда при септооптической дисплазии выявляются те или иные нарушения зрения и эндокринные расстройства, обусловленные пороками развития гипофиза. Из-за этого возможно возникновение вторичных патологий, вызванных аномальной функцией желез внутренней секреции.
Симптомы септооптической дисплазии
Возраст появления симптомов септооптической дисплазии сильно варьирует у разных больных, в тяжелых случаях диагноз может быть поставлен в первые дни и месяцы жизни, при стертой форме заболевания – лишь в младшем или даже старшем детском возрасте. Обычно первым проявлением патологии становится развитие горизонтального нистагма, обусловленное гипоплазией зрительного нерва. Еще раньше при осмотре ребенка могут определяться признаки эндокринной недостаточности гипофиза: гипогликемия, уменьшенный размер половых органов, аномальная желтуха. В редких случаях в первые месяцы жизни при септооптической дисплазии возникают судорожные припадки, длительное сохранение транзиторных рефлексов и другие неврологические нарушения.
По мере роста ребенка, страдающего септооптической дисплазией, может выявляться отставание как в физическом, так и в интеллектуальном развитии. Патологии зрения нарастают, нередко случаются эпилептические припадки. У таких больных обычно раньше, чем у сверстников, начинается процесс полового созревания, обусловленный эндокринными расстройствами. Однако в ряде случаев эндокринные нарушения могут быть выражены довольно слабо или совсем отсутствовать. Такая же ситуация и с задержкой психического развитием больных септооптической дисплазией – она колеблется от нормального интеллекта до глубокой умственной отсталости. Последняя может указывать на наличие сопутствующих пороков развития головного мозга – голопрозэнцефалии, гипоплазии мозолистого тела. Непостоянство симптомов и различная степень их выраженности значительно осложняет диагностику септооптической дисплазии.
Диагностика и лечение септооптической дисплазии
Для определения септооптической дисплазии используются результаты общего осмотра больного, неврологические и офтальмологические исследования, магнитно-резонансная и компьютерная томография, молекулярно-генетические анализы. При осмотре может обнаруживаться отставание в физическом развитии (у детей раннего возраста), раннее наступление полового созревания (у подростков), признаки множественной гормональной недостаточности. Офтальмологическое обследование может выявить нистагм и признаки гипоплазии зрительного нерва – уменьшение размеров диска зрительного нерва, ослабление всех пиков на ЭРГ, полную слепоту. Однако по клинической картине диагностировать септооптическую дисплазию достаточно проблематично, поскольку это состояние характеризуется значительной вариабельностью симптомов.
На магнитно-резонансной томографии определяется отсутствие или выраженное недоразвитие прозрачной перегородки, аплазия или гипоплазия зрительного нерва, в ряде случаев – недоразвитие мозолистого тела. Выявляются нарушения в формировании гипофиза, в особенно тяжелых случаях септооптической дисплазии могут обнаруживаться другие пороки центральной нервной системы, например, голопрозэнцефалия. Биохимический анализ крови позволяет подтвердить наличие недостаточности гормона роста (соматотропина) и других гормонов гипофиза. Молекулярно-генетическая диагностика осуществляется врачом-генетиком, производится прямое секвенирование гена HESX1 с целью подтверждения мутаций. Отсутствие генетических дефектов не является поводом для исключения септооптической дисплазии, так как заболевание может возникать вследствие причин ненаследственного характера. «Золотым стандартом» в диагностике этого состояния являются данные МРТ и КТ.
Специфического лечения септооптической дисплазии не существует, применяют симптоматические и паллиативные лечебные мероприятия. При наличии эндокринных нарушений назначают заместительную терапию, схема которой зависит от характера гормональной дисфункции, что определяется в рамках анализа крови. В большинстве случаев септооптической дисплазии нарушения зрения практически не поддаются коррекции. Симптоматическая терапия включает в себя применение противосудорожных препаратов (при эпилептических припадках) и ноотропных средств, работу детских психологов с больным ребенком при умственной отсталости.
Прогноз и профилактика септооптической дисплазии
Прогноз септооптической дисплазии чаще всего неопределенный из-за сильной вариабельности проявлений заболевания. При наличии тяжелой гипоплазии зрительных нервов, гипофиза, прозрачной перегородки существует крайне высокий риск летального исхода в раннем детстве из-за многочисленных нарушений. Аналогично ухудшают прогноз септооптической дисплазии сопутствующие патологии – детский церебральный паралич, голопрозэнцефалия. Во многих случаях заболевание протекает достаточно благоприятно – симптомы ограничиваются незначительными нарушениями зрения, иногда наличием слабо выраженной умственной отсталости, повышенным риском судорожных припадков. Профилактика септооптической дисплазии возможна только в отношении форм заболевания, обусловленных ненаследственными причинами – инфекционными болезнями матери, ее молодым (юным) возрастом, сосудистыми нарушениями во время беременности.
Оптическая нейропатия Лебера ( Атрофия зрительного нерва Лебера )
МКБ-10

Заболевание названо в честь немецкого офтальмолога Теодора Лебера, который впервые описал 15 случаев внезапной потери зрения у пациентов из четырех семей. Молекулярно-генетические основы заболевания были установлены американским биохимиком Д. Уоллесом в 1988 году. Наследственная оптическая нейропатия Лебера (НОНЛ) встречается с частотой 1 случай на 50 тыс. населения, однако носителями мутации является каждый десятитысячный житель планеты. В России заболевание чаще встречается среди жителей Сибири. Мужчины болеют оптической нейропатией в 5 раз чаще, чем женщины.
Причины
Заболевание развивается вследствие мутаций в митохондриальной ДНК, которые приводят к нарушению энергообеспечения зрительных нервов и вызывают их гибель. До 95% случаев связано с 3 видами генетических аномалий: 3460G>A в гене ND1, 11778GC (ND6). Мутация гена ND4 встречается наиболее часто в клинической практике. Оптическая нейропатия Лебера может быть вызвана и другими вариантами аномалий, которые недостаточно изучены из-за их редкости.
Для мутаций митохондриальной ДНК характерна неполная пенетрантность, поэтому у одних людей с аномальным геном возникает яркая клиническая картина, а другие всю жизнь остаются бессимптомными носителями. В развитии заболевания играют роль внешние триггеры. Наиболее значимыми из них признаны:
- курение;
- воздействие производственных токсинов;
- лекарственные препараты (противотуберкулезные антибиотики, глюкокортикостероиды, интерфероны).
Риск манифестации нейропатии повышается после ЧМТ, сильного стресса, острого соматического заболевания.
При нарушении последовательности нуклеотидов в митохондриальной ДНК изменяется структура белков, которые кодируются данными генами. Поскольку при оптической нейропатии Лебера поражаются ND-гены, кодирующие протеины комплекса I (NADH-убихинон редуктаза), патология возникает на этапе образования молекул АТФ. Клетки нервной системы чувствительны к недостатку энергетических молекул, что и обуславливает поражение зрительного нерва.
Молекулярной основой заболевания считается снижение транспорта АТФ к дистальным участкам аксонов, из-за чего на периферии запускаются процессы апоптоза. Нехватка АТФ наиболее заметна в тонких безмиелиновых волокнах, которые составляют зрительный нерв, поэтому при нейропатии Лебера первично страдает зрение. Развитию патологии способствует врожденный избыток аксонов в диске зрительного нерва и особое строение решетчатой пластины.
Симптомы
В течении оптической нейропатии Лебера выделяют 3 последовательные стадии: доклиническую, острую, хроническую (атрофическую). Первая стадия протекает бессимптомно, однако при обследовании пациента у офтальмолога по другому поводу обнаруживают отек зрительного нерва и появление на нем телеангиэктазий. Длительность этого этапа не регламентирована, поскольку болезнь крайне редко выявляется на доклинической стадии.
Острая стадия оптической нейропатии Лебера чаще всего возникает у молодых мужчин в возрасте от 18 до 30 лет. Пациенты жалуются на резкое снижение зрения по типу центральной скотомы. В течение 1-1,5 месяцев человек утрачивает способность различать мелкие предметы, иногда недоступен даже счет пальцев у лица, и остается лишь светоощущение. Патология поражает оба глаза одновременно или последовательно с интервалом 6-8 недель.
Спустя 6 месяцев болезнь переходит в хроническую стадию, когда острота зрения составляет несколько тысячных. В этот период продолжается атрофия нерва, после завершения которой функцию глазного яблока уже невозможно вернуть. Офтальмологическая картина представлена побледнением зрительного диска. Однако существуют случаи обратного развития симптоматики, когда спустя время пациенты частично восстанавливают зрение.
При оптической нейропатии возможны экстраокулярные проявления. Поражение нервов может затрагивать не только зрительный тракт, поэтому у части пациентов наблюдается периферическая нейропатия, миопатия, мышечная дистония. При манифестации болезни в раннем детском возрасте есть риск развития подострой некротизирующей энцефаломиопатии (синдрома Лея). Изредка наблюдаются перекрестные симптомы НОНЛ и MELAS-синдрома.
Основная проблема нейропатии Лебера заключается в утрате зрения, которая особенно тяжело переносится молодыми больными. На фоне слепоты развиваются тяжелые депрессии, которые могут завершаться суицидальными попытками. Поскольку даже частичное восстановление зрения происходит не у всех, в будущем пациенты получают инвалидность и вынуждены проходить реабилитацию для адаптации к новым условиям жизни.
У некоторых женщин нейропатия Лебера протекает по типу рассеянного склероза с чередованием периодов обострений и ремиссий. У таких пациентов потеря зрения чередуется с эпизодами неполного восстановления способности видеть. Характерно вторичное прогрессирование неврологических симптомов в сочетании со слепотой, известное как болезнь Хардинга. Патология сопровождается очагами демиелинизации в головном мозге, которые отягощают клиническое течение.
Прогрессирующее снижение зрения – повод для всестороннего обследования пациента у врача-офтальмолога. Диагностически значимыми критериями являются: центральная скотома, отсутствие болевого синдрома, наличие подобных симптомов у ближайших родственников по материнской линии. Чтобы подтвердить оптическую нейропатию Лебера, используют следующие исследования:
- Офтальмоскопия. При осмотре глазного дна определяется отек и восковидная бледность зрительного диска. Экскавация центральной части диска в пределах нормы. При обследовании на раннем этапе заболевания визуализируются точечные кровоизлияния в сетчатку.
- Визометрия. Оценка остроты зрения по стандартным таблицам невозможна, поскольку этот показатель не превышает 0,001. На практике это соответствует способности «счета пальцев у лица» и различения силуэтов. Дополнительно проводят периметрию, которая подтверждает выпадение центральных полей зрения.
- Оптическая когерентная томография. Прицельное исследование макулы сетчатки показывает истончение и слабую дифференцировку всех слоев, сглаживание фовеолярного контура, уплотнение внутренней пограничной мембраны. ОКТ – наиболее информативный способ диагностики атрофии ЗН.
- Исследование ЗВП. Электрофизиологическая диагностика зрительных вызванных потенциалов показывает снижение проводимости по нерву на участке до перекреста. Выраженность этих изменений коррелирует со степенью снижения зрения.
- Молекулярно-генетическое тестирование. Выделение митохондриальной мутации необходимо для верификации диагноза нейропатии Лебера. Пациентам назначают таргетное тестирование на 3 самые распространенные точечные аномалии, по показаниям проводят мультигенную панель или полное секвенирование мтДНК.
В остром периоде НОНЛ необходимо отличать от воспалительных заболеваний зрительного нерва: ретробульбарного неврита, оптикомиелита Девика, оптического неврита при ревматических заболеваниях. Обязательно исключают ишемическую оптическую нейропатию, типичную для повышения внутриглазного давления. Хроническую стадию болезни дифференцируют с компрессией зрительного нерва опухолями орбиты и хиазмально-селлярной области.
Лечение оптической нейропатии Лебера
Консервативная терапия
В практической офтальмологии нет эффективной схемы лечения, которая могла бы улучшить зрительную функцию пациентов. В качестве вспомогательной и патогенетической терапии назначаются препараты коэнзима Q10, левокарнитина и цитохрома. Зачастую их комбинируют в «митохондриальные коктейли» для усиления лечебного действия. Однако даже длительный прием медикаментов не оказывает существенного влияния на клинические показатели.
Экспериментальное лечение
Поскольку медикаментозная терапия не позволяет восстановить зрение, ученые возлагают большие надежды на генно-инженерные технологии. Их суть основана на изменении мутантной митохондриальной ДНК на нормальную, чтобы повысить содержание АТФ в аксонах зрительного пути и предотвратить атрофию нерва. На сегодня такие методы находятся на стадии экспериментов, некоторые проходят первую стадию клинических испытаний.
Реабилитация
Для людей с инвалидностью по зрению первостепенную важность приобретает социально-психологическая адаптация. Программа реабилитации предполагает обучение навыкам самостоятельного передвижения, ориентации в пространстве и самообслуживания. Все пациенты проходят изучение шрифта Брайля, что позволяет им читать и продолжать образование. По желанию человека проводится трудовая реабилитации и обучение навыкам (резьба, музыка, скульптура).
Прогностически благоприятным считается НОНЛ с мутацией 14484T>C, при которой пациенты имеют шансы на полное восстановление зрения. Мутация 3460G>A протекает крайне неблагоприятно и быстро приводит к слепоте. Аномалия 11778G
1. Наследственная оптическая нейропатия Лебера/ И.О. Мазунин, Н.В. Володько// Вестник офтальмологии. – 2018. – №2.
2. Harding-синдром-наследственная оптическая нейропатия Лебера и рассеянный склероз: клинический случай и обзор литературы/ Е.В. Попова, В.В. Брюхов, М.В. Коротенкова// Международный неврологический журнал. – 2017. – №7.
3. Оптическая нейропатия Лебера/ Е.А. Руина, О.И. Чадаева, Е.В. Паршина, А.А. Смирнов// Медицинский альманах. – 2017. – №5.
4. Наследственная оптическая нейропатия Лебера/ С.В. Копишинская, С.Н. Светозаров, А.В. Густов// Современные технологии медицины. – 2014. – №2.
Читайте также:
- Лучевая диагностика метастазов в околоушную железу
- Гематология: Лейкоцитарная формула в норме и при болезнях
- Показания, подготовка к артроскопическому релизу и удлинению латерального удерживателя надколенника
- Показания для операции при огнестрельном ранении позвоночника
- Закладка сосудов эмбриона. Формирование аллантоиса эмбриона