Сопутствующие болезни синдрома Тричера-Коллинза в отоларингологии
Добавил пользователь Morpheus Обновлено: 27.01.2026
Атрезией наружного слухового прохода называют отсутствие слухового канала, это состояние может сопровождаться недоразвитием структур среднего и внутреннего уха, вызывая потерю слуха.
Нормальный слух подразумевает функционирование трех областей:
- НАРУЖНОЕ УХО: наружный слуховой проход
- СРЕДНЕЕ УХО: барабанная перепонка и ушные косточки: наковальня, молоточек и стремечко.
- ВНУТРЕННЕЕ УХО: улитка, полукружные каналы, и ушной нерв
При атрезии чаще нарушены наружное и среднее ухо, и большинство детей имеют нормально функционирующее внутреннее ухо (улитку и слуховой нерв).
Атрезии слухового прохода часто сопутствует дефект ушной раковины (микротия), и она так же может быть одно- или двусторонней.
Нарушение слуха при наличии у ребенка слухового канала может быть связано с неправильным формированием среднего уха вследствие отсутствия барабанной перепонки. Еще одна причина снижения слуха - суженный слуховой канал - это стеноз наружного слухового прохода.
При наличии у ребенка признаков атрезии или стеноза наружного слухового прохода нужно пройти обследование сурдолога для прохождения специализированного тестирования слуха и наблюдаться у оториноларинголога (ЛОР-врача).
Варианты лечения атрезии
- Слуховые устройства костной проводимости
Устройства костной проводимости являются хорошим вариантом лечения для коррекции потери слуха у пациентов с односторонней или двусторонней атрезией. Эти устройства обычно называют аббревиатурой BAHA (bone anchored hearing aid). У младенцев и маленьких детей система BAHA закрепляется на голове с помощью мягкой повязки, и может имплантироваться в кость у детей, как только они достигают 5-летнего возраста. Операция подразумевает размещение титанового абатмента или магнитной пластины в черепе. После заживления к этой конструкции присоединяется внешний звуковой процессор. Звуковой процессор вибрирует и таким образом напрямую стимулирует внутреннее ухо (слуховой) нерв. Это позволяет детям с атрезией уха услышать, так он обходит анатомические аномалии в области наружнего и среднего ухо.
Лечение атрезии представляет собой комплексное хирургическое вмешательство, направленное на восстановление просвета наружного слухового прохода для обеспечения звукопроведения и нормализации слуховой функции у детей с односторонней или двусторонней атрезией.
Целью лечения является пластика наружного слухового прохода, что позволит проходить звуковым волнам во внутреннее ухо. Около 50% детей, рожденных с микротией, являются кандидатами на пластику канала. Тем не менее, этот процент ниже у детей с сопутствующими состояниями, такими как синдром Тричер-Коллинза или гемифациальная микросомия.
При костной атрезии слухового прохода хирургическое лечение показано лишь при наличии нормальных структур барабанной перепонки и внутреннего уха, при этом должны быть и нормальные показатели слуха при тканевом звукопроведении. В противном случае хирургическое лечение лишено смысла.
Кандидаты на пластику должны иметь следующее:
- нормальное функционирование структур внутреннего уха, которые преобразуют звуковые вибрации в нервные импульсы, направляемых в мозг (это определяется с помощью аудиограммы)
- адекватное развитие полости среднего уха: целостность цепи слуховых косточек и возможность их вибрации
- благоприятная проекция лицевого нерва
Аудиограмма и компьютерная томография высокого разрешения помогут определить, насколько ваш ребенок является хорошим кандидатом для лечения атрезии. КТ может быть сделано, как только ваш ребенок достигнет, по крайней мере, трех лет, чтобы обеспечить достаточное развитие костных структур среднего уха.
Важно понимать, что при операции установки силиконового имплантата операции по восстановлению слуха делается в первую очередь во избежание повреждения реконструированного уха. А при операции с реберным хрящом каналопластика проводится после реконструкции уха.
Хронология действий- Руководящий принцип заключается в создании максимально и в начале слушания в обоих ушах, чтобы нормальный мозг и слуховой развитие
Руководящий принцип в ведении ребенка с атрезией и микростией заключается в установлении максимально возможного и раннего слуха в обоих ушах для нормального развития мозга и слуха Следующий хронологический список является ориентиром задач для выполнения в течение первых 3 лет жизни ребенка. Этот список является обощенным и может изменяться в зависимости от потребностей и особенностей развития каждого ребенка.
Проверка слуха – определение слухового статуса в обоих ушах. Определение необходимости установки поверхностных устройств костной проводимости при одностороннем поражении, при двустороннем – в обязательном порядке для начала безоперационного лечения, имплантации устройств костной проводимости с 4-месячного возраста.
Диагностика наличия врожденнных синдромов или ассоциированных состояний. (см. ниже)
При односторонних поражениях проведение мероприятий по улучшению слуха в “здоровом” ушке.
Оценка речи и лингвистической функции – определение соответствия их развития по сравнению с детьми с нормальным слухом.
30 месяцев или более:
КТ сканирование для определения стратегии восстановления слуха – определение КТ-степени атрезии. Важно: при наличии у ребенка холестеатомы КТ проводится до 2,5 лет.
Определение метода лечения микротии (наружное ухо) – от этого зависят сроки и последовательность хирургического лечения.
Реберный трансплантат: операции на слуховом проходе выполняется после завершения всех этапов реконструкции ушной раковины и полного заживления в течение нескольких месяцев. При указанном методе лечения операции проводятся с 9-10 летнего возраста и требует около 4 этапов в течение 3х и более лет (при двустороннем поражении). В этот период слух может быть обеспечен поверхностными устройствами проводимости.
Силикон: в первую очередь выполняется пластика слухового канала, начиная с 3-летнего возраста, затем, спустя 6 и более месяцев, формирование ушной раковины с использование силиконового имплантанта.
Сопутствующие болезни синдрома Тричера-Коллинза в отоларингологии
Сопутствующие болезни синдрома Тричера-Коллинза в отоларингологии
Частота встречаемости синдрома Тричера Коллинза составляет один случай на 25-50 тыс. живорождений. Тип наследования — аутосомно-доминантный с различной пенетрантностью. Причиной в 57% случав являются спонтанные генные мутации. Нарушается процесс нормального формирования клеток нервного гребня, что приводит к дефектам развития костной, хрящевой и соединительной тканей.
Классическими признаками синдрома Тричера Коллинза являются гипоплазия нижней челюсти и зубов, ретрогнатия, колобома боковой трети нижних век, отсутствие или недостаток ресничек на верхних веках, антимонголоидный разрез глаз. Аномалии наружного и среднего уха встречаются у 85% пациентов. В легких случаях заболевания единственными видимыми признаками могут быть лишь незначительное опущение скуловой кости и косой разрез глаз с незначительно опущенными углами.
В некоторых случаях диагностика возможна только с использованием КТ, на которой определяется нарушение формирования зубов и нижней челюсти.
У детей с синдромом Тричера Коллинза особо внимание стоит уделять состоянию дыхательных путей. У 47% детей при рождении развивается обструкция дыхательных путей. В большинстве случаев достаточно консервативного лечения без трахеотомии. Для того, чтобы обеспечить проходимость дыхательных путей, нарушенной за счет наличия ретрогнатии и гипоплазии нижней челюсти, обычно достаточно дыхательной маски, носоглоточного воздуховода или положения лежа лицом вниз. В 11% случаев встречается атрезия хоан, усугубляя нарушения дыхания.
Ретроспективный анализ 30 детей с синдромом Тричера Коллинза показал, что только 13% из них в младенчестве потребовалась трахеотомия, в более старшем возрасте экстренная трахеотомия была проведена 7% детей. В одном более раннем и менее крупном исследовании приводились данные о том, что пациентам с челюстно-лицевым дизостозом трахеотомия потребовалась в 41% случаев (к ним относились дети с синдромом Тричера Коллинза и синдромом Нагера).

а - Классическими признаками синдрома Тричера Коллинза являются гипоплазия нижней челюсти с ретрогнатией,
деформации ушных раковин, опущение уголков глаз вниз.
б - Легкие формы синдрома Тричера Коллинза могут проявляться лишь небольшим опущением скул и уголков глаз книзу.
Часто встречается синдром обструктивного апноэ во сне. Для диагностики и определения исходной тяжести заболевания используется полисомнография. У большинства детей с синдромом Тричера Коллинза СОАС разрешается после проведения аденотонзиллэктомии, но некоторым детям может потребоваться длительная СИПАП-терапия или, в тяжелых случаях, трахеотомия. У детей, перенесших трахеотомию по поводу СОАС, эффективным является выдвижение нижней челюсти с дистракционным остеогенезом, после которого пациентов зачастую можно деканулировать (операцию можно проводить в возрасте до 10 лет).
Атрезия и стеноз хоан требуют ранних диагностики и лечения. В случае неэффективности всех предыдущих методов лечения у детей старше 12 лет возможно выполнение выдвижной гениопластики или фиксации подъязычной кости.
Дети с синдромом Тричера Коллинза часто страдают от кондуктивной тугоухости. В исследовании 46 случаев синдрома, было установлено, что степень тугоухости коррелирует с выраженностью деформации ушных раковин. Пациентов стоит информировать о преимуществах и рисках хирургической реконструкции ушных раковин, и в качестве альтернативного лечения предлагать слухопротезирование для костной проводимости или традиционные слуховые аппараты. Вероятность успешного оперативного вмешательства по поводу микротии у детей с синдромом Тричера Коллинза ниже, чем у пациентов с несиндромальной микротией. Тем не менее, для развития речевых навыков мероприятия по улучшению слуха должны проводиться в первый год жизни.
Артикуляция и речь вызывают затруднения у 75% детей с синдромом Тричера Коллинза. Из-за уменьшения размеров носоглотки и ротоглотки у многих детей отмечается гипоназальность. В то же время, примерно у трети детей имеется расщепление неба или небно-глоточная недостаточность, которые вызывают гнусавость. Своевременное оперативное лечение по поводу расщепления неба у таких детей представляет большую сложность из-за наличия сопутствующей обструкции дыхательных путей. Средний возраст детей при проведении уранопластики составлял 2,1 года. Проблемы с артикуляцией также вызваны нарушением прикуса и западением языка.
При синдроме Тричера Коллинза требуется многоэтапное хирургическое лечение. Наибольшую сложность представляет коррекция малярной гипоплазии из-за полной или частичной резорбции трансплантатов. Ко-лобома век устраняется при помощи транспозиционного лоскута, который переносится с верхнего века на нижнее. Коррекция опущенного угла век устраняется выполнением латеральной кантотомии и использованием костных трансплантатов; обычно она проводится одновременно с реконструкцией маляров. В подростковом возрасте с целью устранения горба спинки носа выполняется ринопластика.
Следует формировать у пациента и родителей реалистичные ожидания об успешности операции. Многим пациентам для достижения приемлемых результатов потребуются повторные хирургические вмешательства, иногда остаточные косметические нарушения невозможно устранить даже с помощью максимально интенсивного хирургического лечения.
При правильном ведении командой специалистов разного профиля пациенты с синдромом Тричера Коллинза способны полноценно жить в обществе. Когнитивные нарушения обычно не развиваются. Для достижения наилучших результатов таким пациентам требуется ранняя диагностика, контроль функции дыхания, продуманное хирургическое лечение, а также речевая и психологическая терапия.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма
Профессиональные диагностические инструменты. Оценка эластичности тканей, расширенные возможности 3D/4D/5D сканирования, классификатор BI-RADS, опции для экспертных кардиологических исследований.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
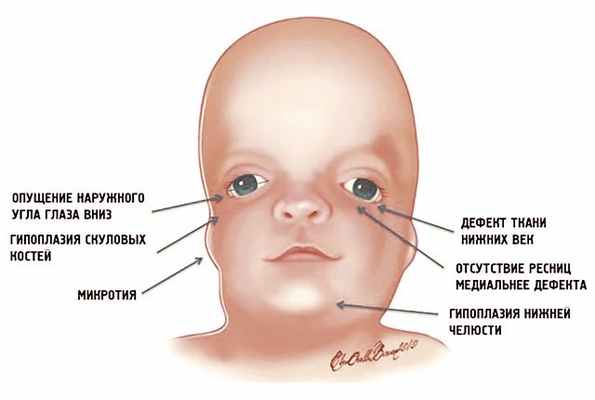
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши – все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности – редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).

Рис. 2. Микрогнатия - сагиттальный скан в 2D, беременность 13 нед.
Полная реконструкция ушной раковины: как проходит лечение микротии в Израиле?
Лечение микротии - одна из самых сложных реконструктивных задач, которая, помимо эстетической функции, призвана решить проблему слуха. Полная реконструкция ушной раковины может также понадобиться пациентам после травмы потери уха, рваной раны или сильного ожога.
- Что такое микротия?
- Причины микротии
- Почему большинство пациентов выбирают лечение микротии ушной раковины за границей
- Методы лечения врожденной аномалии уха у детей в Израиле
- Диагностика аномалий уха в Израиле в клинике Сорока – 100% точность за 3 дня
- Реабилитационный период после операции
- Противопоказания к операции
- Цены на лечение микротии в Израиле в клинике Сорока
- Особенности и классификация стадий микротии
- Чем опасна микротия ушной раковины, если не лечить?
- Отзывы о лечении микротии в Израиле в клинике Сорока
Что такое микротия?
Название заболевания “микротия” происходит от слов «микро» (маленький) и «отия» (ухо). Это врожденное состояние, при котором внешняя часть уха ребенка не развивается должным образом. Микротия бывает:
- односторонней (чаще поражается правое ухо) - 90% случаев;
- двусторонней - у 10%.
Это достаточно редкая аномалия, встречающаяся лишь у 1 из 6 –12 тысяч новорожденных (преимущественно мальчиков), хотя эти показатели могут варьироваться в зависимости от этнического происхождения.
У большинства детей с микротией нет других медицинских проблем, однако эта патология зачастую соседствует с еще одним врожденным слуховым дефектом - атрезией, при которой полностью отсутствует наружный слуховой проход. При этом внутреннее ухо обычно совершенно нормально, но отсутствие естественного ушного отверстия мешает звуку его достигать - в итоге ребенок теряет слух.
Сочетание микротии и атрезии также чаще всего возможно в одностороннем порядке, хотя может выявляться и с обеих сторон. В некоторых случаях слуховой канал снаружи может выглядеть нормально, то есть как бы без микротии, но при этом заканчиваться внутренним «тупиком».
Причины микротии
Было предложено несколько теорий, объясняющих причину микротии во время развития плода. Микротия может возникать изолированно, на ее развитие влияют следующие причины:
- Влияние наследственных (генетических) факторов - доказано лишь в 5% случаев;
- Прием некоторых лекарственных препаратов в первом триместре беременности, например, талидомида и аккутана (изотретиноина);
- Снижение интенсивности кровоснабжения и уровня кислорода в период закладки и развития уха плода;
- Фетальный алкогольный синдром - употребление матерью во время беременности спиртных напитков;
- Употребление наркотиков (метамфетамина) во время беременности.
В большинстве случаев точные причины так и остаются неясными, представляя собой случайный, одноразовый порок развития. Также существует микротия синдромальная или имеющая взаимосвязь с другими врожденными дефектами, среди которых:
- “заячья губа” или “расщепленное нёбо”;
- гемифациальная микросомия;
- синдром Голденхара;
- синдром Тричера-Коллинза и прочие черепно-лицевые синдромы.
Почему большинство пациентов выбирают лечение микротии ушной раковины за границей
При лечении микротии немаловажным будет не только восстановить ушную раковину, но и вернуть слух.
Рассматривая различные варианты, многие предпочитают доверить свое здоровье зарубежным клиникам с целым рядом преимуществ в лечении микротии:
- Опытные реконструктивные пластические хирурги с мировым именем, среди которых - профессор Эльдад Зильберштейн, который занимается лечением в Израиле;
- Новые методы и процедуры, современная аппаратура;
- Щадящие оперативные методики;
- Лояльная ценовая политика: отсутствие предоплаты, возможность оплаты частями с использованием различных платежных форм;
- С иностранными пациентами работают международные отделы при клиниках, помогая в организации поездки, обустройстве быта и оказывая полную поддержку 24 часа в сутки, в том числе и неязыковую.
Методы лечения врожденной аномалии уха у детей в Израиле
При лечении микротии немаловажными факторами остаются не только эстетическая составляющая, но и возвращение естественных слуховых функций организма. Поэтому если есть возможность восстановления слуха, эту операцию проводят первой.
Виды операций по восстановлению слуха:
Имплантация слухового аппарата
Если естественное восстановление слуха невозможно, врач использует специальный имплантат с костной фиксацией. Имплант представляет собой высокопроизводительный звуковой процессор, прикрепляемый к титановой пластине. Его размещают за пораженным ухом, интегрируя с костью черепа. Данный имплант не вживляется детям до 5 лет, однако до наступления операбельного возраста устройство можно крепить к голове с помощью мягкой эластичной повязки.
Атрезиопластика
Эта операция показана для лечения микротии, совмещенной с атрезией, и призвана восстановить нормальный слух путем открытия и формирования ушного канала. Достаточно сложная хирургическая процедура, выполняемая хирургом-отологом.
Операция, связанная с коррекцией формы ушей, называется отопластикой. При этом она имеет 2 направления:
В отопластике применяются сложные технологии, выбор которых обусловлен стадией заболевания и сопутствующими осложнениями. Для улучшения внешнего вида уха используются следующие виды хирургии:
Реконструкция реберного трансплантата - аутогенная реконструкция
Эта процедура представляет собой "выращивание" уха из собственных тканей пациента. Для этой цели хирург вырезает из реберного хряща фрагменты уха, производя полноценный трансплантат, который затем покрывается лоскутом кожи.
Ухо, созданное из собственных тканей ребенка, является живым и растет вместе с ним, оставаясь естественной частью тела. Данный вид операции не проводится у детей до 6 лет - это связано с недостаточным количеством реберного хряща. Однако некоторые более опытные хирурги могут подходить к этому вопросу индивидуально. Скульптура реберного хряща - это поистине хирургическое искусство и технически сложная задача, связанная также с моделированием определенной схожести с противоположным ухом. Следует учесть и тот факт, что, несмотря на то, что у ребенка после удаления реберного хряща остается шрам в области груди, использование собственных тканей в реконструкции уха считается золотым стандартом. Этот метод наиболее безопасен и доступен по стоимости лечения в Израиле.
Реконструкция с использованием искусственного каркаса (Medpor Reconstruction)
Эта технология также предусматривает моделирование уха из собственной ткани и кожи ребенка, однако в качестве каркаса используется безопасный синтетический полиэтилен. Данный тип реконструкции исключает травмирование межреберных хрящей и может проводиться с 2,5-3-летнего возраста. Так как ухо не будет продолжать расти с ребенком, то при моделировании оно выполняется на несколько миллиметров больше по сравнению со здоровым ухом ребенка. Это делается, чтобы со временем обычное ухо естественным путем догнало по размеру реконструированное. Первым в Израиле этот метод внедрил профессор Эльдад Зильберштейн и на сегодняшний день он наиболее опытный пластический хирург в этой технологии.
Протез ушей (искусственное ухо)
В некоторых случаях, если реконструкция невозможна, используется протезирование. Протезы изготавливаются из мягкого и прочного силикона. Слепок для воспроизведения берется со второго уха и окрашивается вручную с максимальным соответствием тону кожи пациента. Протезы крепятся с помощью специального клея, зажимов или магнитов, а также путем остеоинтеграции - крепления на титановые пластины, имплантируемые в кость на естественном месте уха. Возрастные ограничения для остеоинтеграции - от 4 лет. Кости черепа ребенка должны достаточно окрепнуть для операции.
У съемных ушных протезов присутствуют существенные недостатки, по сравнению с имплантированными:
- протез нуждается в регулярной замене;
- его нужно снимать на период сна;
- во избежание попадания инфекций необходимо поддерживать тщательную гигиену кожи в месте крепления.
Поэтапная реконструкция ушной раковины в клинике Сорока
Реконструкция ушной раковины – сложный, длительный и деликатный процесс, происходящий в несколько этапов. Зачастую пациенту необходимы несколько операций с интервалом в 4-6 месяцев, в ходе которых уху придается естественный вид. В общей сложности длительность лечения зависит от степени микротии и может занять больше года.
В клинике Сорока реконструкция выполняется в 4 ключевых этапа, при этом их последовательность и техника выполнения могут варьироваться:
В качестве исходного материала могут использоваться собственные или донорские хрящевые ткани, фрагмент здорового уха или ребра, а также искусственные материалы (полиамидные волокна, силикон и прочее). Преимущества искусственных имплантатов - в сокращении продолжительности процедур, а недостатки - в большой вероятности отторжения. Наиболее предпочтительным будет использование собственных хрящевых тканей пациента.
- Формирование подкожного кармана и ушного блока
На место несформированной или отсутствующей ушной раковины устанавливается каркас, который приживается на протяжении 4-6 месяцев.
Из прижившегося ранее ушного блока формируется основа в нужной анатомической позиции.
С помощью кожно-хрящевого элемента, взятого с заушной области здорового уха, производится реконструкция козелка. Образовавшийся в здоровом ухе дефект скрывается кожной складкой. Продолжительность процесса - 4-6 месяцев.
Хирург, берущийся за подобную реконструкцию, должен быть достаточно опытным и признанным специалистом в области лечения микротии. В клинике Сорока таким специалистом является профессор Эльдад Зильберштейн. Благодаря нему многие дети не только избавились от неэстетичной аномалии, но и начали слышать.
Диагностика аномалий уха в Израиле в клинике Сорока – 100% точность за 3 дня
Микротия редко выявляется при дородовом УЗИ. Обычно любые аномалии в развитии уха или слуха выявляются уже после рождения. Если заболеванию сопутствуют другие патологии, ребенок должен пройти развернутое диагностическое обследование у ряда специалистов. Пациенты с атрезией или стенозом канала обычно не проходят первичную проверку слуха у новорожденных и направляются к детскому аудиологу для дальнейшей диагностической оценки и обсуждения вариантов слуховой реабилитации.
В клинике Сорока предоставляется возможность пройти диагностику в течение 3 дней по предварительной записи:
1-й день – консультация ЛОР-врача
Ведущий ЛОР-врач осматривает пациента, изучает историю болезни: степень развития патологии, возможные причины и назначает необходимые процедуры по обследованию.
2-й день – прохождение диагностики
В список обследования входят:
3-й день – заключение специалиста
Врачи-отоларингологи проводят консилиум с ЛОР-хирургами, для постановки окончательного диагноза по результатам обследования, а также составляют индивидуальную программу решения медицинской проблемы.
В клинике Сорока так же быстро и качественно проходит любая диагностика, в том числе и предшествующая лечению рака в Израиле.
Реабилитационный период после операции
По отзывам пациентов, операция переносится легко, так же как и восстановительный период. Первые дни после операции возможны боли и отеки в области уха. Определенные неудобства доставляет временная невозможность проведения активного отдыха и спортивных занятий. Также врачи рекомендуют отказаться от посещения бани (сауны) или бассейна.
Рекомендации по проведению успешной послеоперационной реабилитации:
- Если операция выполнена маленькому ребенку, то требуется повышенный контроль во избежание нежелательного инфицирования и повреждения прооперированной поверхности.
- К легкой деятельности можно вернуться через 3 дня после операции, а к работе - после снятия швов.
- Первые 7 дней носить повязку со специальной заживляющей мазью. Затем в течение месяца надевать эту повязку на время сна, также крайне важно не спать на оперированном ухе.
- К концу первой недели снимают внешние швы, тогда как внутренние рассасываются сами.
- Необходимо учитывать дополнительный дискомфорт и необходимость восстановления, если в качестве каркаса был взят реберный хрящ и кожный трансплантат пациента.
- В некоторых случаях врач может рекомендовать прием обезболивающих и противоинфекционных препаратов.
Спустя 3 месяца рубцы становятся практически незаметными, отечность и изменение цвета проходят - ухо приобретает нормальный вид. Однако на протяжении всей жизни необходимо обращаться с ним как можно бережнее, оберегая от травм и ушибов.
Следует учитывать, что в процессе восстановления у некоторых пациентов могут проявиться послеоперационные осложнения:
- асимметрия ушей;
- перекос трансплантата.
Возникшие осложнения не являются поводом для повышенного беспокойства и легко решаются после несложной корректирующей операции.
Противопоказания к операции
Ограничения к отопластике аналогичны любым другим при хирургических вмешательствах, однако есть и специфические противопоказания:
- наличие в анамнезе заболеваний, поражающих мелкую капиллярную сеть (болезнь Рейно, сахарный диабет, эндартериит и прочие);
- период обострения хронических болезней;
- наличие у пациента онкологических, инфекционных, сердечно-сосудистых и вирусных заболеваний;
- присутствие кожной сыпи вокруг ушей;
- пониженная свертываемость крови;
- период менструации и беременность;
- некоторые операции имеют возрастные ограничения у детей.
Цены на лечение микротии в Израиле в клинике Сорока
В Израиле цены на лечение микротии у детей на 30-50% ниже, чем в клиниках других стран, клиника Сорока здесь не исключение. Для получения точной информации по ценам и лечению можно провести заочную консультацию по скайпу с ведущим специалистом в этой области профессором Эльдадом Зильберштейном в присутствии медицинского переводчика. Для более продуктивной консультации необходимо подготовить результаты обследования (список можно получить у консультанта на сайте клиники). После консультации профессор представляет письменное заключение о рекомендуемой операции. Стоимость заочной консультации: от $345. Консультация хирурга в клинике: от $398 до $598.
Стоимость операции в клинике Сорока методом Медпор - от $25000 и включает в себя:
- диагностику перед операцией;
- консультацию профессора;
- операцию;
- 1 день госпитализации;
- сопровождение пациента на всех этапах, включая услуги переводчика.
Предоплата не требуется. На сегодняшний день данная операция в Израиле стоит в 2 раза меньшле аналогичной в США.
Особенности и классификация стадий микротии
Микротия встречается во многих вариациях. В некоторых случаях ушной канал очень мал (ушной стеноз) или отсутствует (ушная атрезия). В международной практике принято классифицировать микротию по 4 степеням:
- I степень- неполное развитие уха (или размеры его меньше нормы) с узнаваемыми структурами и небольшим, но присутствующим ушным каналом. Не связана с глухотой.
- II степень- частичная ушная раковина (чаще недоразвитой остается верхняя часть) с закрытым (стенотическим) слуховым каналом. Вызывает кондуктивную глухоту.
- III степень- микротия лобулярного типа. Вызывает проводящую глухоту. Ушной канал и барабанная перепонка обычно полностью отсутствуют (атрезия уха). Ушная раковина тоже отсутствует, однако сохранился небольшой рудимент на месте мочки, имеющий схожесть с арахисом. Это наиболее распространенный тип.
- IV степень - ухо и слух отсутствуют полностью - атония. Встречается достаточно редко.
Профессор Эльдад Зильберштейн из клиники Сорока известен в Израиле и за его пределами своим опытом в лечении всех четырех стадий микротии.
Чем опасна микротия ушной раковины, если не лечить?
Дети с любой стадией микротии, как правило, стесняются своей внешности и подвергаются издевательствам в кругу сверстников. Однако наиболее очевидной проблемой остается снижение слуха в результате сужения или отсутствия ушного канала. Даже при втором здоровом ухе регистрируется планомерное снижение слуха, затрудняющее распознавание отдельных звуков в шумной обстановке. У детей со временем происходит стойкое ухудшение речи.
Отзывы о лечении микротии в Израиле в клинике Сорока
"Наш сыночек родился с микротией 3-й степени правого уха. Мы очень переживали, та к как хоть второе ушко и было здоровым, слух начал постепенно ухудшаться. Наш лечащий врач посоветовал не ждать 6 лет, а уже сейчас ехать в Израиль на лечение. В клинике Сорока мы обратились к профессору Зильберштейну, так как он один может оперировать деток с 3 лет. Сейчас мы на промежуточном этапе лечения, но наш сыночек уже стал слышать на оба ушка. Это такое счастье!"
Синдром обструктивного апноэ сна у детей
Синдром обструктивного апноэ сна (СОАС) — это состояние, проявляющееся повторными кратковременными эпизодами частичной или полной обструкции (непроходимости) верхних дыхательных путей во время сна.
СОАС — остановка дыхания на 3–20 секунд и более — сопровождается частыми ночными пробуждениями, падением уровня кислорода в крови, увеличением уровня углекислого газа, приводит к нарушению качества сна и связанным с этим дневным симптомам.
СОАС может привести к проблемам с обучением, поведением и развитием ребенка. Сердечно-сосудистые, метаболические и нейрокогнитивные сопутствующие заболевания при отсутствии лечения СОАС могут возникать как у детей, так и у взрослых.
Частота СОАС в детской популяции варьируется от 1% до 5% и максимальна в возрастной группе от 2 до 8 лет.
Причины
Во время сна наши мышцы расслабляются, в том числе мышцы задней стенки глотки, помогающие держать дыхательные пути открытыми. При СОАС эти мышцы могут расслабиться настолько сильно, что блокируют дыхательные пути, делая вдох невозможным. Человек делает 1–3 неудачных попытки вдоха, после чего вынужден изменить положение головы, напрячь мышцы, частично или полностью проснуться и с громким храпом наконец сделать вдох. Это приводит к заметному снижению качества сна. Аналогичная ситуация у детей с гипертрофированными (увеличенными) миндалинами или аденоидами, которые могут блокировать дыхательные пути во время сна.
Менее значимыми причинами являются:
- аллергический ринит, образования в полости носа, стеноз носоглотки, искривление перегородки носа, гипертрофия носовых миндалин и др.; ;
- синдром Дауна;
- мукополисахаридоз;
- ахондроплазия;
- макроглоссия (большой язык);
- неврологические нарушения (церебральный паралич, миопатии);
- аномалии челюстно-лицевой области (синдромы Тричера Коллинза, Пьера Робена, Рубенштейна Тейби).
Симптомы
10 признаков ночного апноэ сна, на которые стоит обратить внимание родителям невысыпающихся детей.
1. Храп
Эпизоды храпа возможны при ОРВИ или обострении аллергического ринита у детей с увеличенными миндалинами или аденоидами, но после выздоровления или перехода в ремиссию эти симптомы должны прекратиться. Наличие хронического храпа у ребенка — повод для обращения к врачу: педиатру или оториноларингологу.
2. Частый длительный скрежет зубов (бруксизм)
Повреждение моляров и гипертонус жевательных мыщц — два малоизвестных признака, встречаемых у детей и взрослых с апноэ во сне.
3. Хроническое ротовое дыхание у детей
Носовое дыхание является физиологическим процессом. Нос с его реснитчатым эпителием выполняет барьерную функцию организма, так как при носовом дыхании воздух согревается, увлажняется, частично очищается от пыли и микроорганизмов.
4. Дневная сонливость
Днем дети старше 2 лет редко спят больше одного раза, школьники вовсе не нуждаются в дневном сне при полноценном ночном.
5. Сомнамбулизм (хождение во сне)
По статистике, по ночам «лунатят» от 20% до 30% детей — не менее 1 раза в возрасте от трех до 10 лет. Пик лунатизма приходится на возраст 5 лет и становится менее частым в подростковом возрасте. Эти эпизоды чаще всего возникают в первой трети ночи. Точная причина лунатизма неизвестна, но дети с ночным апноэ чаще других детей бродят во сне.
6. Беспокойный сон
Затруднение дыхания при СОАС может проявляться чрезмерными движениями во время сна. Часто это первый признак апноэ во сне у детей младшего возраста, у которых еще не полностью сформировались зубы. Дети с апноэ во сне часто спят в необычных позах. Их можно найти в кровати «вверх ногами», параллельно изголовью или в другой необычной позе, облегчающей дыхание.
7. Избыточная потливость (гипергидроз)
Если ребенок не болеет ОРВИ, в квартире поддерживается оптимальный микроклимат (температура воздуха 18–22 ˚C, влажность 40–60%), скорее всего нужно искать причину на уровне верхних дыхательных путей.
8. Непроизвольное мочеиспускание ночью (энурез)
Энурез наблюдается у 3–30% детей в возрасте от 4 до 12 лет. Непроизвольное мочеиспускание часто происходит в фазе медленного сна, когда ребенок крепко спит, и его мочевой пузырь наполнен, и также может косвенно указывать на СОАС.
9. Отставание в физическом развитии
Одной из причин физического развития ребенка ниже среднего уровня может быть нарушенный ночной сон, особенно его медленные фазы. Оценку физического развития ребенка педиатр проводит на приеме с помощью центильных таблиц.
10. СДВГ
Синдром дефицита внимания и гиперактивности — поведенческое расстройство, начинающееся в детском возрасте. Проявляется такими симптомами, как трудности концентрации внимания, гиперактивность и плохо управляемая импульсивность.
СДВГ является довольно распространенным диагнозом у детей и часто ассоциирован с нарушениями сна. Апноэ во сне часто упускают из виду у детей с СДВГ, что приводит к неправильной диагностике и неадекватному лечению.
Диагностика
Диагноз в первую очередь ставится клинически и подтверждается полисомнографическими данными.
Альтернативные методы обследования, которые не заменяют полисомнографию, но могут применяться:
- анкетирование;
- ночная пульсоксиметрия;
- мониторирование сна;
- видеоанализ сна;
- периферическая артериальная тонометрия (одобрена FDA c 12 лет, не подходит детям младше 6 лет). Из недостатков: неспособность аппарата различать типы респираторных эпизодов (обструктивное/смешанное/центральное апноэ/гипопноэ);
- слип-эндоскопия;
- биомаркеры (калликреин, уромодулин, урокортин, орозомукоид).
Лечение синдрома обструктивного апноэ сна у детей
Лечение зависит от возраста ребенка, клинических и анамнестических данных, результатов полисомнографии, а также наличия или отсутствия обструкции верхних дыхательных путей, обычно вторичной по отношению к увеличенным аденоидам и/или миндалинам, аллергическому и неаллергическому риниту, острому и хроническому синуситу и другой патологии верхних дыхательных путей. При наличии увеличенных аденоидов или миндалин (или обоих состояний) ребенок нуждается в хирургическом лечении: частичном или полном удалении миндалин и аденоидов. В 20–40% случаев СОАС после удаления миндалин и аденоидов сохраняется; чаще это происходит в возрасте старше 7 лет, при тяжелой степени апноэ, у пациентов с бронхиальной астмой и пациентов негроидной расы.
При неудаче хирургического лечения (или отказе от него) также может быть применена симптоматическая лекарственная терапия (например, назальные стероиды при аллергическом рините) или терапия «сипап» (СРАР).
СРАР (Continuous Positive Airway Pressure) — постоянное положительное давление в дыхательных путях — метод лечения СОАС с помощью специального аппарата, всасывающего очищенный, нагретый, увлажненный при помощи фильтра комнатный воздух, подающего этот воздух под специальным давлением через назальную маску. То есть человек надевает герметично прилегающую маску на лицо и ложится с ней спать.
- раздражение кожи, слизистой оболочки полости носа, конъюнктивы;
- плохая переносимость;
- деформация челюстно-лицевой области.
Также при СОАС заметное облегчение может принести изменение образа жизни (снижение веса, отказ от вредных привычек, налаживание режима сна) — особенно людям с избыточным весом или ожирением, чтобы терапевтический эффект СРАР был максимальным.
Читайте также:
- Функциональные пробы при венозной гипертонии. Фармакологические пробы при венозной гипертонии
- ЭКГ при предсердной экстрасистоле
- Субарахноидальное кровоизлияние при алкоголизме. Клиника субарахноидального алкогольного кровоизлияния
- МРТ травматической эпидуральной гематомы
- Ангиосаркома при лимфостазе. Внутрисосудистая эндотелиома.