Течение бета-талассемии (анемии Кули) - осложнения
Добавил пользователь Алексей Ф. Обновлено: 29.01.2026
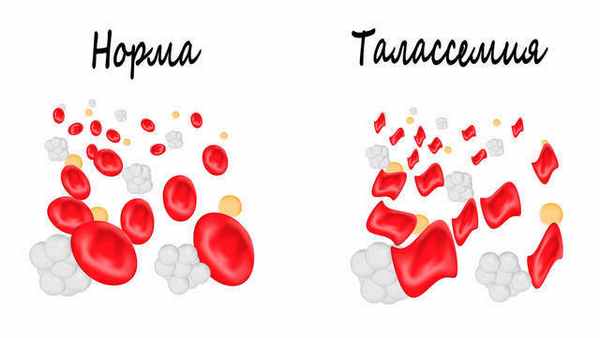
Талассемия (анемия Кули) – наследственное заболевание, при котором нарушается синтез (выработка) гемоглобина. Генетическая патология чаще встречается у народов Азии, Ближнего и Среднего Востока, афроамериканцев, редкие случаи болезни регистрируются в России на территории Поволжья. Клинические проявления разнообразны и зависят от формы талассемии. Иногда заболевание диагностируется в первые дни жизни, в других случаях долго протекает бессимптомно. Больные с легкими формами живут полной жизнью и не нуждаются в лекарственной терапии.
Содержание
Классификация
Талассемию классифицируют по тяжести заболевания, типу наследования и виду генетического дефекта. Подробная классификация с пояснениями представлена ниже в таблице.
| Признак классификации | Форма заболевания | Комментарии |
| Вид мутации | альфа-талассемия | связана с мутациями в генах HBA1 и HBA2; при нарушениях в одном гене протекает бессимптомно, изменения двух или трех генов вызывают хроническую анемию, мутации четырех генов несовместимы с жизнью |
| бета-талассемия | выделяют большую и малую бета-талассемии; малая форма никак не проявляется, большая форма характеризуется тяжелым течением | |
| Тип наследования | гетерозиготная | развивается при наследовании одного измененного гена |
| гомозиготная | развивается при мутации у обоих родителей | |
| Тяжесть заболевания | легкая средней степени тяжелая | степень тяжести заболевания зависит от количества мутировавших генов; при легких формах симптомы отсутствуют, тяжелые формы часто осложняются нарушениями кровообращения и приводят к развитию сердечно-сосудистых заболеваний |
Талассемия чаще встречается в регионах, где распространена малярия. Исследователи связывают это с тем, что носители данного вида мутаций в генах более устойчивы к малярийному плазмодию.
Причины заболевания
Причина развития болезни – мутация в гене, который несет информацию о синтезе цепей глобина – белка, участвующего в образовании гемоглобина . Вследствие чего его продуцирование (выработка) прекращается либо происходит в меньшем количестве, клетки крови разрушаются, и развивается анемия. При гомозиготной форме талассемии характерно наличие двух дефектных генов, которые наследуются от обоих родителей. При гетерозиготном варианте человек становится носителем измененного гена, унаследованного от одного из родителей.
Патогенез
Молекулярную основу патогенеза талассемии составляет генетический дефект, который заключается в том, что в клетках у одних пациентов функционирует аномальная РНК (рибонуклеиновая кислота, участвует в кодировании, прочтении и регуляции генов), а у других происходит делеция генетического материала (изменение в структуре хромосом). Вследствие этого снижается или прекращается синтез одной из цепочек гемоглобина. В норме синтез цепей гемоглобина сбалансирован, а количество альфа- и не альфа-цепей одинаково. Изменение синтеза одной из них приводит к нарушению баланса. Так, недостаток бета-цепей при сохранении продукции альфа-цепей приводит к избытку свободных альфа-цепей, и наоборот.
Симптомы талассемии
Клиническая картина талассемии зависит от формы заболевания. Однако для большинства пациентов характерны следующие признаки :
- задержка роста и развития;
- изменение цвета кожи (бледность, желтушность, потемнение);
- склонность к переломам из-за истончения костей;
- деформации скелета;
- увеличение печени и селезенки;
- камни в желчных путях.
При тяжелой форме бета-талассемии клинические проявления заметны уже в течение первого года жизни ребенка . Характерные признаки этой формы заболевания:
- монголоидное лицо (уплощенная переносица, сужение глазных щелей);
- измененная форма головы («башенный череп»);
- увеличенная верхняя челюсть;
- неправильный прикус.
Малая форма бета-талассемии протекает бессимптомно, и выявляется при помощи методов лабораторной диагностики.
Обратите внимание! Талассемия может привести к умственному и физическому недоразвитию ребенка, поэтому заболевание нельзя игнорировать. При подозрении на генетическую патологию нужно обязательно пройти обследование, и при подтверждении диагноза начать лечение.
Диагностика заболевания
Патологию можно заподозрить у детей с характерными клиническими признаками, в роду которых были выявлены случаи талассемии. Пациентам с подозрением на генетическую мутацию необходимо посетить врача-гематолога (специалиста по болезням крови) и медицинского генетика. Подтвердить или опровергнуть диагноз поможет лабораторная диагностика :
- Типичными признаками талассемии в анализе крови служат снижение уровня гемоглобина и цветового показателя, наличие эритроцитов измененной формы, повышение показателей железа и непрямого билирубина.
- При исследовании вещества костного мозга обнаруживается гиперплазия (разрастание) красного кроветворного ростка с высоким числом эритробластов и нормобластов (незрелых клеток крови).
- Молекулярно-генетические исследования позволяют выявить мутацию альфа- или бета-глобина, нарушающую синтез цепочек белка глобина.
- При ультразвуковом исследовании (УЗИ) брюшной полости выявляется увеличение печени и селезенки, обнаруживаются камни в желчном пузыре.
Дифференциальная диагностика заболевания проводится с железодефицитной, аутоиммунной, серповидно-клеточной и другими видами анемий.
Лечение талассемии
Лечение талассемии зависит от формы заболевания. Больные с малой бета-талассемией не нуждаются в лекарственной терапии, кроме приема витаминов группы B и фолиевой кислоты. Пациентам с гомозиготной бета-талассемией с первых дней жизни показано проведение гемотрансфузионной терапии (переливание эритроцитарной массы), введение хелатирующих препаратов (для связывания железа), гормонов при резком ухудшении состояния. В тяжелых случаях больным с талассемией показана трансплантация костного мозга.
Осложнения заболевания
При тяжелых формах заболевание может приводить к развитию осложнений. Среди наиболее частых осложнений генетической мутации белка глобина встречаются :
- Повышение уровня железа в крови. Избыток железа в организме возникает вследствие прогрессирования заболевания либо в результате частых переливаний крови. Высокий уровень железа приводит к повреждению печени, эндокринной системы, становится причиной развития аритмии.
- Увеличение селезенки (спленомегалия). Данная генетическая мутация часто сопровождается разрушением большого числа эритроцитов, создавая повышенную нагрузку на селезенку, вследствие чего орган увеличивается в размерах. Спленомегалия может усугубить течение анемии и снизить продолжительность жизни. При большом увеличении селезенки может потребоваться хирургическая операция по её удалению – спленэктомия. Оперативное вмешательство рекомендуется проводить пациентам старше шести лет.
- Инфекционные осложнения. При генетической мутации повышается вероятность развития инфекционных заболеваний. Наиболее высокий риск отмечается у пациентов после удаления селезенки.
- Деформация костей. Талассемия может привести к разрастанию костного мозга и деформации костей. Увеличение костного мозга также способствует истончению костей и повышению вероятности переломов.
Последствия заболевания
Прогноз при тяжелых мутациях неблагоприятный, высока вероятность летального исхода. Больные умирают в детстве или молодом возрасте, редко доживают до тридцати лет. При малой бессимптомной форме продолжительность и качество жизни не страдают. Заболевание не оказывает негативных последствий на организм. Больным достаточно регулярно обследоваться, соблюдать рекомендации врача и придерживаться здорового образа жизни.
Профилактика талассемических мутаций
Для предупреждения риска рождения детей с талассемией родителям необходимо еще на этапе планирования беременности посетить медицинского генетика . Особенно это актуально для семей, имеющих в роду генетические мутации той или иной степени выраженности. Современная антенатальная диагностика с использованием высокотехнологичного оборудования позволяет выявить большинство талассемических мутаций.
Если талассемия выявлена уже после рождения ребенка, важно вовремя начать ее лечение. Своевременная адекватная терапия сведет риск развития осложнений и костных деформаций у ребенка к минимуму.
Талассемия
Талассемия, в буквальном переводе "морская анемия", возникает она при мутации генов, отвечающих за синтез пептидов, из которых состоит гемоглобин, и тяжесть заболевания зависит от числа изменённых генов. Особенность этой анемии в том, что пациенту приходится бороться с избыточным потреблением железа.
Что такое талассемия и почему она возникает
Талассемия – это генетическое заболевание крови. Образуется недостаточное количество гемоглобина, белка, отвечающего за перенос кислорода и углекислого газа. Он состоит из четырех пептидов. 90% молекул гемоглобина содержат два альфа-глобина и два бета-глобина (гемоглобин А или A1), 2.5% содержат вместо бета-цепей дельта-цепи (гемоглобин А2), а остальные представляют собой гемоглобин A, состарившийся в процессе эксплуатации в эритроцитах (гемоглобин A3). При мутации в генах, отвечающих за синтез одного из глобинов, состав гемоглобина нарушается. Эти изменения влекут гибель эритроцитов.
Причины и факторы риска развития заболевания
Талассемия – заболевание наследственное, то есть мутация передается от родителя ребенку. Если кто-то из родственников страдал от этой патологии, риск развития заболевания повышается.
Второй фактор риска – этническая принадлежность. Больше всего талассемия распространена в Африке, Средней Азии и странах Средиземноморья, где она и была открыта (в переводе с греческого “талассемия” - это “морская анемия”). Различают два основных типа талассемии: альфа и бета. В первом случае мутация затрагивает гены, отвечающие за синтез альфа-глобинов, во втором – бета.
Альфа-глобины кодируются четырьмя генами. Тяжесть заболевания будет зависеть от количества патологически измененных участков ДНК:
- 1 измененный ген – бессимптомная форма. Но при ней человек становится носителем заболевания и может передать его своим детям;
- 2 гена – легкое течение болезни;
- 3 гена – тяжелое течение болезни;
- 4 гена – редкий тип заболевания, который плохо совместим с жизнью. Большинство плодов гибнет еще в период внутриутробного развития, а родившиеся малыши, в основном, умирают вскоре после рождения или нуждаются в пожизненной терапии. В отдельных случаях удается их вылечить путем пересадки костного мозга.
Бета-глобины кодируются одним геном, который локализуется в 11-й хромосоме. Если дефектный ген содержится только в одной хромосоме из пары, заболевание протекает легко (малая талассемия). Повреждение обеих хромосом приводит к очень серьезному заболеванию, известному как большая талассемия или болезнь Кули (анемия Кули).
Коды МКБ-10 (Международная классификация болезней 10 пересмотра):
- альфа-талассемия - D56.0
- бета-талассемия - D56.1
- дельта-бета-талассемия - D56.2
- носительство признака талассемии - D56.3
- наследственное персистирование фетального гемоглобина (НПФГ) - D56.4
- другие талассемии - D56.8
- талассемия неуточненная - D56.9
Симптомы и признаки талассемии
В большинстве случаев талассемию определяют еще на этапе дородовой диагностики. При необходимости лечение начинают сразу, не дожидаясь появления симптомов. Если заболевание не выявила пренатальная диагностика, ожидаются следующие симптомы:
- бледность или желтушность слизистых оболочек;
- замедленный рост;
- темная моча;
- увеличение живота;
- деформация костей, особенно костей черепа.
Время появления первых признаков талассемии во многом зависит от типа заболевания и количества мутаций. У одних детей симптомы регистрируются вскоре после рождения, у других – в первые два года жизни.
Диагностика талассемии
Симптомы при талассемии бывают более или менее характерными. Чтобы поставить окончательный диагноз, врачу необходимы результаты лабораторных исследований. Обязателен при подозрении на талассемию общий анализ крови. Он покажет сниженное количество эритроцитов мелких, светлых, разных по форме и размеру. Кроме зрелых клеток, в мазке будет немало их предшественников - бластов. Дополнительно могут назначить другие специфические анализы крови для определения степени тяжести нарушений (биохимический анализ, определение железосвязывающей способности плазмы или ферритина в сыворотке). Также разработаны молекулярные тесты (ПЦР), позволяющие определить наличие мутаций.
Для оценки состояния печени, селезенки используют УЗИ, а для выявления патологии костной ткани – рентгенографию.
У ребенка талассемия может быть диагностирована еще на этапе вынашивания. Это исследование особенно рекомендуется проводить родителям, которые больны или могут быть носителями этого заболевания. Существует два метода диагностики:
- биопсия ворсинок хориона – проводится на 11-ой неделе беременности;
- амниоцентез (отбор околоплодных вод) – назначают на 16-й неделе.
Лечение талассемии
Определяется типом и степенью тяжести. При умеренно выраженных симптомах лечение не назначают. Время от времени проводят только переливание крови. В основном это нужно после операций, родов или для предотвращения возможных осложнений. Люди с бета-талассемией требуют более частых переливаний крови. Для нормализации избыточного уровня железа им также назначают специфические препараты, которые связывают и выводят железо.
При выраженной и тяжелой формах болезни существует два способа лечения:
- частые переливания крови (раз в несколько недель), которые сочетают с приемом препаратов, выводящих лишнее железо из организма;
- пересадка костного мозга – единственный метод, который помогает полностью излечить человека от талассемии. К сожалению, далеко не всегда трансплантация бывает успешной.
Лекарственные препараты при талассемии назначают только для коррекции симптомов и осложнений. Медикаментозной терапии самого заболевания не существует.
Осложнения талассемии
Возможные осложнения заболевания:
- излишек железа, которое входит в состав гемоглобина. Накопление этого элемента в организме приводит к поражению сердца, печени, эндокринной системы;
- подверженность инфекциям. Особенно актуально для пациентов, которым удалили селезенку;
- деформация костей, связанная с увеличением объема костного мозга. Чаще всего этот процесс затрагивает кости черепа, реже конечностей. Они истончаются и чаще ломаются;
- спленомегалия - увеличение селезенки, где в основном и гибнут дефектные эритроциты. Если селезенка увеличена очень сильно, ее удаляют. Эта операция называется спленэктомия;
- задержка роста и полового созревания;
- сердечные болезни (хроническая сердечная недостаточность и аритмии) могут развиться при тяжелом течении заболевания.
Правильный образ жизни при заболевании
Существуют простые советы, которые помогают людям с талассемией лучше переносить симптомы заболевания.
- не употреблять никакие витаминно-минеральные комплексы или пищевые добавки, содержащие железо;
- сбалансированное разнообразное питание;
- профилактика инфекционных заболеваний, в первую очередь вакцинация.
Прогноз при талассемии
При легкой форме талассемии прогноз очень хороший. Такие пациенты не требуют постоянного лечения, и осложнения крайне редки. При средней и тяжелой форме прогноз также хороший, но необходимы регулярные переливания крови и прием препаратов, связывающих железо. Такие лекарства очень важны, поскольку наибольшее количество смертей больных талассемией связано с накоплением этого элемента в организме. Пациентам с костными осложнениями может понадобиться операция.
Профилактика талассемии
Талассемия – заболевание наследственное. Поэтому, если один или оба родителя больны, необходимо проконсультироваться с врачом-генетиком на этапе планирования беременности.
Талассемии
(средиземноморская анемия; большая и малая талассемия)
, MD, PhD, Johns Hopkins University School of Medicine
- Патофизиология
- Клинические проявления
- Диагностика
- Прогноз
- Лечение
- Основные положения
- Дополнительная информация
Талассемии – это группа врожденных микроцитарных гемолитических анемий, которые характеризуются дефектом синтеза гемоглобина. Альфа-талассемия особенно распространена среди лиц африканского, средиземноморского, или южноазиатского происхождения. Бета-талассемия более распространена у лиц средиземноморского, ближневосточного, южноазиатского и индийского происхождения. Симптомы и признаки обусловлены анемией, гемолизом, спленомегалией, гиперплазией костного мозга, при многократных гемотрансфузиях может наблюдаться перегрузка железом. Диагностика основана на генетическом исследовании и количественном анализе структуры гемоглобина. Лечение тяжелых форм может включать в себя гемотрансфузии, спленэктомию, терапии хелаторами и трансплантацию стволовых клеток.
Патофизиология талассемий
Талассемия – гемоглобинопатия Обзор гемоглобинопатий Гемоглобинопатии – это генетические нарушения, влияющие на структуру или синтез молекулы гемоглобина. Молекулы гемоглобина состоят из полипептидных цепей, химическая структура которых контролируется. Прочитайте дополнительные сведения , которая является одним из наиболее распространенных наследственных заболеваний, связанных с синтезом гемоглобина. Нормальная зрелая молекула гемоглобина (гемоглобин А) состоит из 2 пар цепей, называемых альфа и бета. Нормальная кровь взрослого человека также содержит ≤ 2,5% Hb A2 (состоит из альфа- и дельта-цепей) и 2% гемоглобина F (фетального гемоглобина), который имеет гамма-цепи вместо бета-цепей. Талассемия является результатом несбалансированного синтеза гемоглобина, вызванного снижением синтеза по крайней мере одной полипептидной цепи глобина (бета, альфа, гамма, дельта).
Альфа-талассемия
Альфа-талассемия является результатом снижения синтеза альфа-полипептидных цепей вследствие делеции одного или нескольких генов альфа-цепей. Люди, как правило, имеют четыре гена альфа-цепей (по два на каждой паре хромосом), потому что ген альфа-цепей дублируется. Классификация болезни основана на количестве и местоположении делеций:
Альфа + талассемия: Потеря одного гена на одной хромосоме (альфа/--)
Альфа 0 талассемии: Потеря обоих генов на одной и той же хромосоме (--/--)
Бета-талассемия
Бета-талассемия вызвана снижением синтеза бета-полипептидных цепей в результате либо мутации, либо делеции в гене бета-глобина, что приводит к нарушению синтеза гемоглобина А. Мутации или делеции могут привести к частичной потере (бета + аллель) или полной потере (бета 0 аллель) функции бета-глобина. Существуют два гена бета-глобина, и у пациентов могут быть гетерозиготные, гомозиготные или сложные гетерозиготные мутации. Кроме того, пациенты могут быть гетерозиготными или гомозиготными по аномалиям в 2-х различных генах глобина (например, бета и дельта).
Бета-дельта-талассемия является менее распространенной формой бета-талассемии, при которой нарушается синтез как дельта-цепи, так и бета-цепи. Эти мутации могут быть гетерозиготными или гомозиготными.
Симптомы и признаки талассемий
Клинические особенности талассемий сходны, но различаются по степени тяжести, в зависимости от количества нормального гемоглобина.
Альфа-талассемия
Пациенты с одной альфа + аллелью (альфа/альфа; альфа/--) являются клинически нормальными и называются бессимптомными носителями.
У гетерозигот с дефектами в 2 из 4 генов, таких как две альфа + аллели (альфа/--; альфа/--) или 1 альфа 0 аллель (альфа/альфа; --/--) наблюдается тенденция к развитию микроцитарной анемии легкой или умеренной степени тяжести, но с субклиническим течением. Данные пациенты имеют малую альфа-талассемию.
Дефекты в 3 из 4 генов, вызванные совместным наследованием как альфа +, так и альфа 0 (альфа/-; -/-), существенно нарушают синтез альфа-цепи. Синтез поврежденных альфа-цепей в результате приводит к образованию тетрамеров избыточных бета-цепей, называемых Hb H, а у младенцев - к формированию гамма-цепей, называемых гемоглобином Барта. У пациентов с болезнью гемоглобина Н часто наблюдаются гемолитическая анемия и спленомегалия.
Дефект всех 4 генов через две альфа 0 аллели (--/--;--/--) является летальным состоянием, которое вызывает внутриутробную гибель плода (водянку плода), поскольку гемоглобин, не содержащий альфа-цепей, не переносит кислород.
Бета-талассемия
При бета-талассемии, клинические фенотипы подразделяются на 3 группы в зависимости от степени нарушения синтеза бета-глобина:
Малая бета-талассемия (характерно) возникает у обычно бессимптомных гетерозигот (бета/бета + бета/бета 0) с клинической картиной микроцитарной анемии от легкой до умеренной степени. Этот фенотип может также возникнуть в легких случаях бета +/бета +.
Промежуточная бета-талассемия проявляется вариабельной клинической картиной, которая является промежуточной между большой и малой талассемией, обусловленная наследованием 2 аллелей бета-талассемии (бета +/бета 0 или, в тяжелых случаях, бета +/бета +).
Большая бета-талассемия (или анемия Кули) возникает у гомозиготных пациентов (бета 0/бета 0) или сложных гетерозигот (бета 0/бета +) в результате тяжелого дефекта бета-глобина. У этих пациентов развивается тяжелая анемия и гиперактивность костного мозга. Большая бета-талассемия проявляется в возрасте от 1 до 2 лет с симптомами тяжелой анемии и трансфузионной и абсорбционной перегрузки железа. У пациентов наблюдаются желтуха, язвы нижних конечностей и холелитиаз (как при серповидноклеточной анемии Серповидно-клеточная анемия Серповидноклеточная анемия ( гемоглобинопатия) является причиной хронической гемолитической анемии, которая наблюдается практически исключительно у представителей негроидной расы. Она вызвана. Прочитайте дополнительные сведенияДиагностика талассемий
При наличии подозрения диагностика данного типа гемолитической анемии включает в себя:
Мазок периферической крови
Исследование структуры ДНК (пренатальная диагностика)
Малую талассемии обычно выявляют при проведении рутинного мазка периферической крови и развернутого анализа крови, когда обнаруживают микроцитарную анемию и повышенное количество эритроцитов. При желании, диагноз малой бета-талассемии может быть подтвержден с помощью количественных исследований структуры гемоглобина. Никакого вмешательства не требуется; у женщин анемия может усугубляться беременностью.
Более тяжелую талассемию следует подозревать у пациентов с отягощенным наследственным анамнезом, при наличии характерных клинических симптомов или микроцитарной гемолитической анемии. При подозрении на талассемию выполняются обычные лабораторные тесты для выявления микроцитарных гемолитических анемий и количественный анализ структуры гемоглобина. Характерно повышение уровня билирубина, железа и ферритина в сыворотке крови.
При альфа-талассемиях процентное содержание Hb F и Hb A2 обычно в пределах нормы, выявление одного или двух дефектных генов, характерных для талассемии, производится с помощью современных генетических тестов. Диагноз часто ставится путём исключения других причин микроцитарной анемии.
При большой бета-талассемии наблюдается тяжелая анемия, часто со снижением уровня гемоглобина ≤ 6 г/дл (≤ 60 гр/л). Количество эритроцитов повышено по отношению к уровню гемоглобина, поскольку наблюдается выраженный микроцитоз. Диагностика основана на исследовании мазка периферической крови, в котором обнаруживается множество ядросодержащих эритробластов, мишеневидных клеток, небольших бледноокрашенных эритроцитов; характерна точечная или диффузная базофилия.
При количественном анализе структуры гемоглобина диагностическим критерием малой бета-талассемии является повышенный уровень Hb A2. При большой бета-талассемии обычно повышено содержание Hb F, иногда до 90%, а содержание Hb A2 обычно > 3%.
Болезнь гемоглобина Н диагностируется по выявлению Hb H или фракций Барта при электрофорезе гемоглобина. Наличие специфического молекулярного дефекта не меняет клинического подхода.
Стандартом пренатальной диагностики и генетического консультирования является картирование генов рекомбинантной ДНК (в частности метод полимеразной цепной реакции [ПЦР]).
Если при анемии выполняется исследование костного мозга (к примеру, для исключения других причин), выявляется выраженная эритроидная гиперплазия.
При визуальном исследовании, выполненном по другим причинам, у пациентов с большой бета-талассемией можно выявить изменения, обусловленные хронической гиперактивностью костного мозга. Наблюдаются истончение кортикального слоя костей черепа, расширение диплоических пространств, лучистая трабекулярная структура, появление гранул или феномен «матового стекла». В трубчатых костях могут наблюдаться очаги остеопороза, расширение костномозгового пространства и истончение кортикального слоя. Тела позвонков могут иметь зернистый внешний вид, или вид матового стекла. Фаланги могут быть прямоугольными или двояковыпуклыми. Визуализация грудной клетки может выявить признаки паравертебрального экстрамедуллярного гемопоэза.
Прогноз при талассемии
Продолжительность жизни у пациентов с малой бета-талассемией или малой альфа-талассемией является нормальной. Прогноз при заболевании Hb Н и промежуточной бета-талассемии вариабельный.
Продолжительность жизни снижена у пациентов с большой бета-талассемией, главным образом в связи с осложнениями в результате хронических трансфузий.
Лечение талассемий
Часто – переливание эритроцитарной массы с/без железохелатирующей терапии
Спленэктомия при наличии спленомегалии
Если возможно, выполняют трансплантацию аллогенных стволовых клеток
Луспатерцепт для лечения трансфузионно-зависимой бета-талассемии
Пациентам с малой альфа-талассемией или малой бета-талассемией лечение не требуется.
При гемоглобинозе Н: спленэктомия может помочь при тяжелой анемии или наличии спленомегалии.
Пациенты с промежуточной бета-талассемией должны получать как можно меньше гемотрансфузий, чтобы избежать перегрузки железом. Тем не менее супрессия патологического гемопоэза с помощью периодических трансфузий эритроцитов Эритроциты (ККТ) Цельная кровь способствует улучшению кислородной емкости крови, расширению объема и замене факторов свертывания, а раньше ее назначали при сильной кровопотере. Однако, поскольку компонентная. Прочитайте дополнительные сведения может быть ценной у пациентов с тяжелым течением заболевания. При большой бета-талассемии следует проводить трансфузии по необходимости для поддержания уровня гемоглобина примерно 9 - 10 г/дл (от 90 до 100 г/л) и избегать развития тяжелых клинических проявлений.
Чтобы предотвратить или отсрочить развитие осложнений от перегрузки железом, должен быть удален избыток (трансфузионного) железа (например, путем назначения длительной железохелаторной терапии Лечение Вторичная перегрузка железом появляется в результате избыточной абсорбции железа, повторяющихся переливаниях крови или избытке перорального приема, как правило, у пациентов с нарушениями эритропоэза. Прочитайте дополнительные сведения ). Хелаторную терапию, как правило, начинают, когда уровни сывороточного ферритина превышают 1000 нг/мл (> 1000 мкг/л) или после приблизительно от 1 до 2 лет проведения плановых трансфузий. Спленэктомия может снизить потребность в гемотрансфузиях у пациентов с выраженной спленомегалией.
Люспатерцепт представляет собой инъекционный рекомбинантный белок слияния, который ингибирует метаболический путь передачи сигналов от трансформирующего фактора роста бета. В рандомизированном плацебо-контролируемом исследовании у пациентов с бета-талассемией он снизил потребность в трансфузии на 33% у 21% пациентов (по сравнению с 4,5% в контрольной группе). Луспатерцепт является вариантом лечения для пациентов, которым необходимо переливание крови ( 1 Справочные материалы по лечению Талассемии – это группа врожденных микроцитарных гемолитических анемий, которые характеризуются дефектом синтеза гемоглобина. Альфа-талассемия особенно распространена среди лиц африканского. Прочитайте дополнительные сведения ).
Справочные материалы по лечению
1. Cappellini MD, Viprakasat V, Taher A, et al: A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med 382(13):1219–1231, 2020. doi: 10.1056/NEJMoa1910182
Основные положения
Талассемия является результатом снижения синтеза по меньшей мере одной полипептидной цепи глобина (бета, альфа, гамма, дельта); в результате аномальные эритроциты являются микроцитарными, часто неправильной формы и склонны к гемолизу (вызывающему анемию).
Часто проявляется спленомегалия, часто массивная, которая может привести к селезеночной секвестрации, ускоряющей разрушение эритроцитов (в том числе введенных при переливании).
Часто наблюдается перегрузка железом из-за увеличенного поглощения (в связи с нарушенным эритропоэзом), а также из-за частых трансфузий.
Диагностика с использованием электрофореза гемоглобина.
Следует проводить трансфузии по необходимости, проводя мониторинг перегрузки железом и используя хелаторную терапию.
Спленэктомия может снизить потребность в гемотрансфузиях у пациентов со спленомегалией.
Трансплантация аллогенных стволовых клеток имеет излечивающий эффект.
Дополнительная информация
Ниже следует англоязычный ресурс, который может быть информативным. Обратите внимание, что The manual не несет ответственности за содержание этого ресурса.
Cooley's Anemia Foundation: provides comprehensive patient education and support and advocacy to patients with thalassemia
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Талассемия (Анемия кули)
Талассемия — это наследственное заболевание, для которого характерно угнетение формирования цепочечных белковых молекул. Они создают структуру гемоглобина, что отражается на повреждении красных клеток, мембраны эритроцитов, формировании гемолитических кризов. На наличие болезни указывает анемический синдром, гепатоспленомегалия, изменения костей. Подтверждение производится только после анализов и обследования.
Под названием заболевания Талассемия подразумевается целая группа болезней крови генетического характера. Распространена проблема в Черноморском и Средиземноморском регионах. Также случаи болезни часто фиксируются в Закавказье, Средней Азии, Индии, Индонезии, на Ближнем Востоке. Ежегодно примерно 300 000 детей по всему миру рождаются с симптомом. Заболевание делится на бессимптомную, тяжелую, легкую и фатальную формы.
Альфа-талассемия, Бета-талассемия
Есть несколько видов поражения, их определяют с учетом пораженной полипептидной цепи:
- А-талассемия. В данном случае подавлению подвергается синтез HbA (альфа-цепи). Включает наличие немого и манифестного генов, водянку плода, также встречается гемоглобинопатия Н;
- Б-талассемия. Характеризуется проблемами с бета-цепями синтеза HbA. Включает анемию Кули, гомозиготное, гетерозиготное поражение.
Талассемия у детей
Сбой синтеза составляющих гемоглобина — наследственная болезнь, она основана на отхождении от нормы в структуре генов, которые влияют на цепное строение глобина. После мутации ген передается от родителей к их потомству. Цепи не образуются, либо их объема недостаточно. Остальные цепочки в глобине формируются, что приводит к нестабильности и порче эритроцитов. Выявляется не только анемия, но и отложение железа в тканях и органах.
У детей череп приближается к форме квадрата, глаза сужаются, а зона переносицы приходит к седловидной конфигурации. Значительно меняется верхняя челюсть, ее размер превышает параметры нижней. Также проявляются язвы в области голеней, кожа становится желтоватой и бледной. Снижается природная защита организма, наблюдается отставание в развитии, утомляемость, вялость, в желчных путях образуются билирубиновые камни. При формировании водянки плода, он обычно умирает до рождения.
Анализы на талассемию
По лабораторным анализам смотрят на:
- Снижение железа и рост эритробластов;
- Уменьшение окрашенности и уровня гемоглобина;
- Присутствие неправильных по форме эритроцитов и прочие показателей.
Симптомы и признаки
Более всего заметные признаки — изменения области головы и увеличение живота, дополненные бледностью и желтоватым оттенком кожи.
- Медленное развитие и рост;
- Изменения скелета;
- Увеличение селезенки, печени;
- Истончение конечностей и частые переломы;
- Неправильный прикус;
- Камни в желчных путях.
Малая форма может быть бессимптомной, ее можно выявить после проведения лабораторных исследований. В ряде случаев наблюдается постоянная усталость, одышка, симптоматика, характерная для интоксикации, темная моча. Возможно недоразвитие (физически и умственно), поэтому требуется лечение.
Диагностика
Врач выполняет общий осмотр и проверяет наследственный фактор, при болезни гемоглобин падает до 30-50 единиц. Также актуальны биохимические исследования, взятие мазка крови, ПЦП диагностика, иногда обязательна пункция костного мозга. В числе необходимых для диагностики шагов числится рентген костей, электрофорез вещества на ацетат-целлюлозной пленке. Устанавливаются размеры желез и внутренних органов, а также проверяется череп на расширения, гранулы, уплотнения или их отсутствие. Во время беременности желательно пройти скрининг с целью исключения генетических патологий.
Носитель талассемии
Носители имеют один ген с дефектом, при этом от самой болезни такие люди не страдают. В данном случае талассемия не развивается, но есть вероятность небольшой анемии, поскольку габариты эритроцитов меньше, чем в случае нормы. Состояние отличается от железодефицитной категории, поскольку в лечении нет необходимости. Для проверки носительства достаточно сдать анализ крови. Любой может быть носителем, но чаще всего поражаются люди, которые произошли из Среднего востока, Пакистана, Бангладеша и ряда других приморских регионов. Людям из группы риска рекомендуется сдать кровь.
Лечение
При малой талассемии лечение не нужно, важно сбалансированное питание, здоровый образ жизни, отсутствие вредных привычек. Также нужно быть активным физически. При среднем или большом поражении требуется лечение:
- Выведение излишков железа из организма (хелатная терапия);
- Переливание эритроцитарной массы;
- Нередко требуется удаление селезенки.
Препараты назначает врач, они нужны для предотвращения ряда осложнений и коррекции симптоматики. Лечения с использованием медикаментов нет. При среднем течении болезни потребность во вливаниях донорских частиц крови возникает при операциях, во время беременности, при резком падении гемоглобина, сердечной недостаточности. Также потребность наблюдается при проблемах опорно-двигательный системы и легочной гипертензии.
Ситуация становится хуже при большой талассемии, поэтому требуется:
- Ежемесячные переливания крови пожизненно;
- При спленомегалии удаляется селезенка;
- Удаление лишнего железа;
- Пересадка стволовых клеток.
После операции по удалению селезенки падает потребность в гемотрансфузиях, одновременно с этим отмечается ухудшение самочувствия по причине высокой вероятности проявления инфекций. Радикальное лечение в тяжелых случаях — пересадка костного мозга, такой подход актуален для взрослых и детей, он позволяет сохранить максимальное качество жизни. Если лечение большой бета-талассемии нет, процесс завершается смертью в раннем возрасте.
Лечение контролируется с помощью УЗИ, сдачи крови на общий анализ, ЭХО-ЭКГ, биохимическим исследований. Серьезное лечение и правильный образ жизни при средней тяжести позволяет получить хорошие прогнозы. Тяжелое течение без комплексного лечение приведет к низкому качеству жизни и ее короткому сроку. Поэтому при наличии заболевания у родственников важно пройти обследование, особенно при планах на рождение ребенка.
Информация носит справочный характер и не является руководством к действию. Не занимайтесь самолечением. При первых симптомах заболевания обратитесь к врачу.
Бета-талассемия

Медицинский эксперт по заболеванию
Проф. Полина СтепенскиРуководитель отделения ТКМ и иммунотерапии у детей и взрослых клиники «Хадасса»
Стаж: более 24 лет
Запись на прием
В клинике «Хадасса» очень успешно лечат бета-талассемию с помощью трансплантации костного мозга. Процедуру проводит ведущий специалист Израиля, заведующая отделением ТКМ и иммунотерапии у детей и взрослых проф. Полина Степенски.
В клинике на высочайшем уровне проводится генетическая диагностика бета-талассемии, она проходит под руководством одного из самых известных генетиков страны — проф. Орли Эльпелег.
Если ребенку не удалось найти донора для ТКМ и было принято решение об использовании стволовых клеток следующего ребенка, специалисты по медицине плода обеспечивают рождение детей без генной мутации посредством процедуры отбора здорового зародыша (PGD) с последующим ЭКО. «Хадасса» по праву гордится своими достижениями в данной области.
Что такое бета-талассемия

На фото брат и сестра из Индии. Девочка страдала от талассемии. Ее здоровый млаший брат был рожден в клинике «Хадасса» с помощью предимплантационной генной диагностики (PGD). В возрасте 1 года он стал донором костного мозга и спас сестру. (2015 год)
Талассемия — группа тяжелых генетических заболеваний крови, при которых нарушается синтез жизненно важного белка гемоглобина, входящего в состав эритроцитов, обеспечивающих кислородом все ткани и органы. Болезнь развивается из-за нарушения синтеза гемоглобина, связанного со сниженной продукцией одной из полипептидных цепей глобина (альфа, бета,гамма, дельта). Если нарушается синтез альфа-глобиновых цепей, болезнь классифицируют как альфа-талассемию. В случае недостаточного синтеза бета-глобиновых пептидных цепей, патологию определяют как бета-талассемию.
Заболеваемость альфа-талассемией распространена в южно-азиатских и западноафриканских странах. Бета-талассемия чаще встречается в Средиземноморских регионах, а также станах Северной Африки западной Азии. Ежегодно по всему миру рождается более 280 тысяч детей с синдромом талассемии.
По данным ВОЗ, бета-талассемия является наиболее распространенной формой из группы наследственных заболеваний крови по аутосомно-рецессивному типу. В равной степени затрагивает как женщин, так и мужчин. Болезнь дифференцируют на гомозиготную форму, когда генетически она наследуется от одного из родителей и гетерозиготную форму, в случае наследования мутантного гена от обоих родителей.
Основная причина возникновения заболевания — мутации гена, который отвечает за синтез полипептидной цепи гемоглобина. Дефект на молекулярном уровне обусловлен неэффективным процессом или мутацией регуляторных генов, соединением аномальной матричной РНК или хромосомные перестройки структурных генов.
Механизм развития бета-талассемии заключается в недостаточном количестве синтезируемых бета-цепей, что приводит к избыточному продуцированию альфа-цепей, которые откладываются в клетках эритроидного ряда и повреждают их. Процесс сопровождается гибелью ретикулоцитов в селезенке, разрушением эритроцитов и деструкцией эритробластов в костном мозге, а также накоплением фетального гемоглобина. В результате этого сокращается транспортировка кислорода по всем тканям и органам, возникает хроническая анемия и тканевая гипоксия, что в свою очередь становится причиной многих заболеваний, в том числе костных деформаций и т.д.
Чтобы получить более подробную информацию или консультацию специалиста по орфанным заболеваниям, заполните все поля формы. Наши консультанты будут рады Вам помочь.
МКБ-10
В класс МКБ-10 входят заболевания крови и кроветворных органов, а также гемолитические анемии, талассемии и тяжелая бета-талассемия (анемия Кули). По международной классификации болезней из группы МКБ-10, бета-талассемии присвоен код D56.1.
Симптомы
Клиническая картина при бета-талассемии проявляется у детей в течение 1-2 лет после рождения. Гомозиготная форма болезни сопровождается слабостью, хронической усталостью, повышением температуры тела, одышкой, бледностью и желтушностью кожного покрова, интоксикацией. Вследствие хронической анемии у детей наблюдается значительная задержка в физическом и умственном развитии. Нередко при бета-талассемии обнаруживаются нарушения в работе сердца, печени, эндокринных желез, что вызвано патологическим накоплением железа в организме. Из-за экстрамедуллярного кроветворения и тотального разрушения эритроцитов увеличивается селезенка и печень.
На фоне поражения трубчатых костей возникают патологические переломы, развивается синовит крупных суставов, желчнокаменная болезнь. Пациенты с бета-талассемией подвержены частым инфекционным болезням. Гомозиготная форма может привести к серьезным осложнениям: кардиосклерозу, сахарному диабету, сердечной недостаточности, циррозу печени, сепсису, пневмонии.
Бета-талассемия гетерозиготной формы может протекать бессимптомно или с незначительными проявлениями: повышенной утомляемостью на фоне гипохромной анемии, головокружениями, небольшим увеличением селезенки, головными болями.
Признаки
Уже в первый год после рождения, у детей с большой бета-талассемией можно отметить внешние признаки заболевания. Это монголоидный овал лица, характерный разрез глаз, четырехугольная (башенная) форма черепа, расширенная носовая перегородка (седловидная переносица), увеличенная верхняя челюсть, неправильный прикус. Наблюдается изменения скелетной структуры, иногда могут образовываться изъязвления в области голеней на фоне желтушности кожи. У маленьких детей достаточно выраженные внешние признаки бета-талассемии, что дает основания для предварительного диагноза.
Диагностика
При подозрениях на бета-талассемию специалисты в первую очередь изучают семейный анамнез, учитывают жалобы пациента и проводят физикальный осмотр. Для постановки диагноза требуется комплексное обследование, состоящее из таких процедур:
- Лабораторные исследования крови, позволяющие определить уровень железа, концентрацию билирубина, средний объем эритроцитов, нарушения цепей гемоглобина и другие показатели.
- Молекулярно-генетические исследования на предмет генных мутаций.
- Пункция костного мозга, подтверждающая гиперплазию красного кроветворного ростка.
- Краниография черепа, выявляющая игольчатый периостоз (симптом волосатого черепа).
- УЗИ брюшной полости — для оценки состояния желчевыводящих путей, а также размеров печени и селезенки.
В процессе обследования бета-талассемию дифференцируют от аутоиммунной, железодефицитной и серповидно-клеточной анемии, а также от наследственного микросфероцитоза. Немаловажное значение имеет пренатальная диагностика. Женщинам, у которых в семье есть родственники с подобным диагнозом, необходимо провести забор амниотической жидкости, чтобы подтвердить или опровергнуть наличие генетической патологии у плода. Данное исследование позволит начать своевременное лечение.
Чтобы получить более подробную информацию или консультацию специалиста по орфанным заболеваниям, заполните все поля формы. Наши консультанты будут рады Вам помочь.
Классификация
В зависимости от генетических факторов и степени тяжести симптоматической картины, бета-талассемию разделяют на 3 группы:
Малая бета-талассемия — легкая форма заболевания, когда полной или частичной мутации подвержен только 1 ген, отвечающий за синтез бета-цепей. В анамнезе у носителей этой формы болезни отмечается лишь небольшой уровень эритроцитов в крови и легкая степень анемии.
Большая бета-талассемия — тяжелая форма патологии, при которой мутация затрагивает оба гена, ответственных за синтез полипептидных цепей гемоглобина. При большой бета-талассемии у детей до года, существует высокий риск летального исхода.
Промежуточная бета-талассемия — отличается большим дефицитом бета-цепей, может переходить в тяжелую форму болезни. В большинстве случаев протекает без серьезных нарушений в организме с минимально выраженной клинической картиной.
Лечение бета-талассемии
Тактика лечения зависит от формы и степени тяжести талассемии. Пациенты с малой бета-талассемией в лечении не нуждаются и могут самостоятельно компенсировать последствия врожденного заболевания, соблюдая здоровый образ жизни (отказ от вредных привычек, двигательная активность, сбалансированное питание).
При большой и промежуточной бета-талассемии врачи применяют следующие методы лечения:
- Трансфузионная терапия — переливание донорской крови. В случае промежуточной формы бета-талассемии трансфузию проводят редко, при сердечной недостаточности, легочной гипертензии, тяжелых костных деформациях, значительной задержке роста. Большая форма заболевания требует регулярного переливания эритроцитов (1-2 раза в месяц).
- Хелаторная терапия — введение препаратов, связывающих железо и выводящих его из организма. При гемолитических кризах используют глюкокортикоиды.
- Спленэктомия — удаление селезенки в случае ее гипертрофии.
- Пересадка костного мозга (ТКМ) — трансплантация донорских стволовых клеток, способствует возобновлению нормальной работы кроветворной системы.
Также пациентам со всеми формами бета-талассемии показан прием витаминов В, С, Е и фолиевой кислоты. Для предотвращения инфекционных осложнений требуется вакцинация.
Прогноз заболевания и профилактика
После пересадки костного мозга многие пациенты полностью выздоравливают. Постоянное переливание эритроцитной массы увеличивает продолжительность жизни маленьким детям, у которых была выявлена большая бета-талассемия. При гетерозиготной форме заболевания с бессимптомным течением качество и продолжительность жизни не страдают.
Профилактика бета-талассемии заключается в планировании беременности будущих родителей. Супружеской паре необходимо обязательно проконсультироваться с генетиком, если у кого-то из родственников была выявлена анемия Кули. При высоком генетическом риске носители мутантных генов должны отказаться от деторождения. Также к первичной профилактике бета-талассемии относят дородовую диагностику плода.
Если вам необходима консультация специалиста по орфанным заболеваниям, заполнив все поля формы, и наши консультанты будут рады Вам помочь.
Читайте также: