Токсические осложнения трансплантации гемопоэтических стволовых клеток (ТГСК)
Добавил пользователь Валентин П. Обновлено: 29.01.2026
Фаза I / II испытания трансплантации гемопоэтических стволовых клеток (ТГСК) для детей с генетическим заболеванием клеток крови без HLA-совместимого родственного донора
Это клиническое испытание трансплантации костного мозга для пациентов с диагнозом генетическое заболевание клеток крови, у которых нет HLA-совместимого родственного донора. Генетический Заболевания клеток крови включают: Дефекты эритроцитов, например. гемоглобинопатии (серповидноклеточная болезнь и талассемия), анемия Блэкфана-Даймонда и врожденная или хроническая гемолитическая анемии; Дефекты лейкоцитов / иммунодефициты, например. хроническая гранулематозная болезнь, синдром Вискотта-Олдрича, остеопетроз, синдром Костманна (врожденная нейтропения), Наследственный лимфогистиоцитоз (HLH); Дефекты тромбоцитов, например, врожденный амегакариоцитарный тромбоцитопения; Нарушения обмена веществ/накопления лейкодистрофии, мукополисахаридозы, Болезнь Гурлера; Дефекты стволовых клеток, например ретикулярная агенезия, среди многих других редких подобных условия. В плане исследуемого лечения используется новый режим лечения трансплантатов, целью которого является снижение острой токсичности и осложнений, связанных со стандартными планами лечения и улучшить результат Стволовые клетки крови будут получены либо от неродственного донора, либо от неродственной пуповины. кровь.
Это пилотное клиническое испытание трансплантации гемопоэтических стволовых клеток у пациентов с диагноз генетического заболевания клеток крови, у которых нет HLA-совместимых братьев и сестер донор. Стволовые клетки будут получены от 1) совместимого неродственного донора (MUD) или 2) неродственного донора. пуповинной крови (ПКК). Пациенты получат новый режим кондиционирования с бусульфаном, Цитоксан и флударабин (Bu/Cy/Flu) и либо алемтузумаб (Campath 1H) для реципиентов MUD или кроличий антитимоцитарный глобулин (rATG) для реципиентов неродственных UCB до трансплантация гемопоэтических стволовых клеток (ТГСК). Предполагается, что уменьшенные дозы цитоксана уменьшат острую токсичность. связаны со стандартной химиотерапией бусульфаном и цитоксаном (т.е. синусоидальным обструктивный синдром (СОС), геморрагический цистит и мукозит). И добавление флударабина к режиму кондиционирования с миелоаблативными дозами бусульфана и сниженным дозы Цитоксана перед ТГСК преодолеют барьер приживления, создаваемый интактным трансплантатом. иммунной системы, что наблюдается у пациентов с генетическим заболеванием.
Тип вмешательства: Процедура
Описание: режим кондиционирования трансплантации гемопоэтических стволовых клеток в зависимости от источника трансплантата
Критерии включения: - Летальные или сублетальные генетические заболевания клеток крови, у которых отсутствуют полностью гистосовместимые родной брат или другой семейный донор - Генетические заболевания, которые могут быть кандидатами для этого протокола, включают те, которые Было показано, что аллогенная ТГСК приносит пользу: дефекты эритроцитов, дефекты лейкоцитов/ Первичные иммунодефициты, Дефекты тромбоцитов, Нарушения обмена веществ/накопления и Стволовые дефекты клеток. - Почки: клиренс креатинина или скорость клубочковой фильтрации (СКФ) ≥50 мл/мин/1,73 м2 и не требующие диализа. - Легочные: FEV1, FVC и DLCO (с поправкой на гемоглобин) ≥ 50% от должного. если не в состоянии выполнить тесты функции легких, затем сатурация O2 ≥ 92% в комнатном воздухе. - Сердечная: фракция выброса левого желудочка в покое должна быть ≥ 40% или укорочение фракция ≥ 26% - Печень: билирубин ≤3x верхняя граница нормы (ULN) и АЛТ и AST ≤5x для возраста (с исключение изолированной гипербилирубинемии вследствие синдрома Жильбера). - Пациенты будут в возрасте от 0 до 21 года. - Критерии включения по конкретному заболеванию (в соответствии с протоколом). Критерий исключения: - Реципиенты не должны иметь каких-либо общих критериев исключения, а также конкретных заболеваний критерии исключения, когда это применимо. - Пациент с гистосовместимым братом или сестрой - Отказ органов-мишеней, препятствующий переносимости процедуры трансплантации, включая режим кондиционирования. - Клиренс креатинина или СКФ < 50 мл/мин/1,73 м2 или почечная недостаточность, требующая диализа. - Врожденный порок сердца, приводящий к застойной сердечной недостаточности. - Тяжелое остаточное заболевание/нарушение ЦНС [(кроме только гемиплегии) например. кома или трудноизлечимые судороги] - Вентиляционная недостаточность - Серьезные врожденные аномалии, отрицательно влияющие на выживаемость, напр. пороки развития ЦНС - по шкале Лански < 40% или по шкале Карновски < 60% - серопозитивность к ВИЧ - Диагностика анемии Фанкони, тяжелого комбинированного иммунодефицита (ТКИД) - Положительный тест на беременность (для женщин в детородном периоде) - Неконтролируемые бактериальные, вирусные или грибковые инфекции (в настоящее время принимает лекарства, но клинические симптомы прогрессируют) - Критерии исключения по конкретному заболеванию (в соответствии с протоколом).
Тип: Главный следователь
Принадлежность следователя: Детская больница Лос-Анджелеса
ФИО следователя: Hisham Abdel-Azim
Должность следователя: Главный исследователь
Описание: Unrelated donor
Описание: Пуповинная кровь
Модель вмешательства: Параллельное назначение
Первичное назначение: Уход
Маскировка: Нет (открытая этикетка)
This information was retrieved directly from the website clinicaltrials.gov without any changes. If you have any requests to change, remove or update your study details, please contact [email protected] . As soon as a change is implemented on clinicaltrials.gov, this will be updated automatically on our website as well.
Трансплантация гемопоэтических стволовых клеток
Трансплантация гемопоэтических стволовых клеток (ГСК) – быстроразвивающаяся технология, которая потенциально может позволить добиться излечения при злокачественных заболеваниях крови ( лейкемиях Обзоры лейкемии (Overview of Leukemia) Лейкоз представляет собой злокачественное заболевание, характеризующееся производством избыточного количества незрелых или аномальных лейкоцитов, что в конечном итоге приводит к подавлению производства. Прочитайте дополнительные сведения , лимфомах Обзор лимфомы (Overview of Lymphoma) Лимфомы представляют собой гетерогенную группу заболеваний, происходящих из клеток ретикулоэндотелиальной и лимфатической системы. Основные варианты лимфом – лимфома Ходжкина и неходжкинские. Прочитайте дополнительные сведения , миеломах Множественная миелома Множественная миелома является злокачественной плазмоклеточной опухолью, продуцирующей моноклональные иммуноглобулины, которые внедряются в прилежащую костную ткань и разрушают ее. К характерным. Прочитайте дополнительные сведения , миелодисплазии Миелодиспластический синдром (МДС) Миелодиспластический синдром (МДС) представляет собой группу заболеваний, характеризующихся цитопенией в периферической крови, дисплазией гемопоэтических клеток-предшественников, гиперклеточностью. Прочитайте дополнительные сведения ). Трансплантация ГСК также иногда используется при солидных опухолях (например, некоторые опухоли зародышевых клеток), которые реагируют на химиотерапию. (См. также Обзор трансплантации (Overview of Transplantation) Обзор трансплантации (Overview of Transplantation) Трансплантатами могут быть собственные ткани пациента (аутотрансплантанты; например, кости, костный мозг и трансплантаты кожи) Генетически идентичная (сингенная [от монозиготных близнецов]). Прочитайте дополнительные сведения ).
Трансплантация ГСК содействует излечению путем:
Восстановления костного мозга после миелоаблативного уничтожения рака
Замены аномального костного мозга на нормальный костный мозг при доброкачественных гематологических заболеваниях
Трансплантация ГСК может быть аутогенной (с использованием собственных клеток пациента) или аллогенной (с использованием клеток от донора). Забор стволовых клеток могут проводится из
Периферическая кровь в значительной степени заменила костный мозг как источник стволовых клеток, особенно при аутотрансплантации ГСК, поскольку проводить забор стволовых клеток легче, а восстановление уровня нейтрофилов и тромбоцитов происходит быстрее. Трансплантация ГСК пуповинной крови зачастую используется только у детей, поскольку для взрослого человека в ней слишком мало стволовых клеток. Потенциальным будущим источником стволовых клеток являются плюрипотентные стволовые клетки (определенные клетки, взятые у взрослых и перепрограммированные, чтобы действовать как стволовые клетки).
Для аутогенной трансплантации ГСК нет противопоказаний.
Относительные противопоказания к аллогенной трансплантации ГСК включают возраст старше 50 лет, наличие предыдущих трансплантаций ГСК (ТГСК) и тяжелые сопутствующие заболевания.
Аллогенная ТГСК ограничена, в основном, нехваткой тканесовместимых доноров. Идеальным донором является брат или сестра с идентичным главным комплексом гистосовместимости человека (НLA), далее следует брат или сестра с совместимым HLA. Поэтому часто используются родственные HLA-несовместимые доноры ГСК или неродственные совместимые (найденные в международных регистрах). Однако, уровень долгосрочной выживаемости без сопутствующих осложнений может быть ниже по сравнению с трансплантациями ГСК от HLA-идентичных родственных доноров.
Технология использования ГСК, выделенных из пуповинной крови, в стадии отработки, находится в зачаточном состоянии, но набирает интерес. Около 20 000 процедур трансплантаций пуповинной крови было сделано с момента введения этой процедуры в 1989 году. Поскольку пуповинная кровь содержит незрелые стволовые клетки, HLA-подбор не так важен, чем при других типах трансплантации гемопоэтических стволовых клеток. Одной из проблем, связанных с процедурой, является незрелая антигенная природа иммунных клеток в пуповинной крови, что приводит к увеличению процента необученных Т-клеток и увеличивает степени риска реактивации инфекций цитомегаловирусом или вирусом Эбштейна-Барр.
Методика
Для выделения костномозговых стволовых клеток аспирируется 700–1 500 мл (максимально 15 мл/кг) костного мозга из заднего гребня подвздошной кости донора; при этом используется местная или общая анестезия.
Для выделения стволовых клеток из периферической крови донору вводят рекомбинантные факторы роста (гранулоцит-колониестимулирующий фактор или гранулоцит-макрофаг-колониестимулирующий фактор) для стимуляции пролиферации и мобилизации стволовых клеток, последующий стандартный аферез проводится через 4–6 дней. Затем для идентификации и выделения стволовых клеток производится флуоресцентно-активированная сортировка клеток.
Стволовые клетки вводятся в течение 1–2 часов посредством центрального венозного катетера большого диаметра.
Режимы кондиционирования
Чтобы не произошло отторжения трансплантата, перед аллогенной трансплантацией гематопоэтических стволовых клеток в случае рака реципиенту для индуцирования ремиссии и супрессии иммунной системы в первую очередь назначают режим кондиционирования (например, миелоаблятивный режим, такой как назначение циклофосфамида 60 мг/кг/день 1 раз в день внутривенно в течение 2 дней с общим полнодозовым облучением всего тела, или бусульфан 1 мг/кг перорально 4 раза в день в течение 4 дней и циклофосфамид без общего облучения).
Подобные режимы кондиционирования используются при аллогенной ТГСК, даже если это при данном злокачественном заболевании не показано, для снижения частоты случаев отторжения и рецидива.
Такие схемы кондиционирования не используется до аутогенной ТГСК при заболевании раком; вместо этого используются специфические к раку лекарственные средства.
Немиелоаблативный иммуносупрессивный режим кондиционирования (например, циклофосфамид, облучение тимуса, антитимоцитарный глобулин [АТГ], и/или циклоспорин) может снизить риск заболевания и смерти и полезен пожилым пациентам, пациентам с сопутствующими заболеваниями и восприимчивым к реакции «трансплантат против опухоли» (например, с множественной миеломой).
Режимы с пониженной интенсивностью (например, флударабин с мелфаланом, пероральным бусульфаном или циклофосфамидом) по степени интенсивности и токсичности находятся между миелоаблативными и немиелоаблативными режимами кондиционирования. Цитопения, возникающая в результате лечения, может быть продолжительной и приводить к серьезным осложнениям и смерти, а также требует назначения терапии стволовыми клетками.
После трансплантации
После трансплантации гемопоэтических стволовых клеток, реципиент получает колониестимулирующие факторы для уменьшения продолжительности посттрансплантационной лейкопении, профилактический курс лекарственных средств Инфекция для защиты от инфекций, а при аллогенной ТКСК – профилактический курс иммуносупрессантов продолжительностью до 6 месяцев (обычно метотрексат и циклоспорин) для предупреждения реакции со стороны донорских Т-лимфоцитов по отношению к молекулам HLA реципиента (болезнь «трансплантат против хозяина»). Если пациент не лихорадит, от приема антибиотиков широкого спектра действия обычно воздерживаются.
Приживление трансплантата обычно происходит через 10–20 дней после ТГСК (раньше в случае трансплантации стволовых клеток из периферической крови) и определяется по абсолютному числу нейтрофилов > 500 мкл ( > 0,5 × 10 9 /л) на литр.
Осложнения трансплантации гемопоэтических стволовых клеток
Осложнения после трансплантации стволовых клеток могут возникнуть в раннем периоде (менее чем через 100 дней после трансплантации) или в позднем периоде. После аллогенной ТГСК риск инфекций увеличивается.
Ранние осложнения
Серьезные ранние осложнения включают:
Острая реакция «трансплантат против хозяина» (РТПХ)
Нарушение приживления и отторжение встречаются у 5% пациентов и проявляются персистирующей панцитопенией или необратимым снижением числа клеток крови. Лечение проводится глюкокортикоидами в течение нескольких недель.
Острая РТПХ отмечается у реципиентов при аллогенной трансплантацией КСК (у 40% пациентов, получивших клетки от HLA-совместимых сибсов, и у 80% – от неродственных доноров). При таком состоянии отмечается лихорадка, сыпь, гепатит с гипербилирубинемией, рвота, диарея, боли в животе (с возможным развитием кишечной непроходимости) и потеря веса.
Факторы риска острой РТПХ включают:
Несоответствие по половому признаку и системе НLA-антигенов
Пожилой возраст реципиента и/или донора
Предварительная сенсибилизация донора
Неадекватная профилактика БТПХ
Диагноз острой БТПХ ставится на основании данных анамнеза, объектвного обследования и результатов печеночных проб. Лечение: метилпреднизолон 2 мг/кг внутривенно 1 раз в день, с увеличением дозы до 10 мг/кг при отсутствии ответа в течение 5 дней.
Поздние осложнения
Серьезные поздние осложнения включают:
Хроническая БТПХ может возникать самостоятельно, развиваться из острой БТПХ или появляться после разрешения острой БТПХ. Хроническая БТПХ начинается обычно через 4–7 месяцев после ТГСК (период может варьировать от 2 месяцев до 2 лет). Хроническая БТПХ наблюдается у реципиентов при аллогенной ТГСК (у 35–50% реципиентов, получивших трансплантаты от HLA-совместимых родственных доноров, 60–70% – от неродственных доноров).
Хроническая БТПХ поражает в первую очередь кожу (например, лихеноидная сыпь, склеротические изменения кожи) и слизистые (например, сухой кератоконъюнктивит, периодонтит, орогенитальные лихеноидные реакции), а также желудочно-кишечный тракт и печень. Основной характеристикой является иммунодефицит; могут развиваться также облитерирующие бронхиолиты, подобные тем, которые развиваются при трансплантации легких. В конечном счете, БТПХ приводит к смерти 20-40% пациентов, у которых она развилась.
Лечение БТПХ, которая влияет на кожу и слизистые, необязательно; при более тяжелых состояниях лечение подобно таковому при острой БТПХ. Используя моноклональные антитела или механическую сепарацию, добиваются истощения Т-лимфоцитов в аллогенном донорском трансплантате, что снижает частоту и тяжесть БТПХ, но это также снижает реакцию «трансплантат против опухоли», что может усилить клеточную пролиферацию, улучшить приживание и снизить частоту рецидивов болезни. Частота рецидивов при использовании аллогенных ГСК выше из-за отсутствия влияния реакции "трансплантат против опухоли", и из-за того, что могут быть трансплантированы циркулирующие опухолевые клетки, случайно собраные вместе со стволовыми клетками. Ex vivo исследуются опухолевые клетки, выделенные перед аутогенной трансплантацией.
У пациентов без хронической БТПХ назначение всех иммуносупрессантов может быть прекращено через 6 месяцев после ТГСК; таким образом, поздние осложнения у этой группы пациентов редки.
Прогноз при трансплантации гемопоэтических стволовых клеток
Прогноз после трансплантации ГСК варьирует в зависимости от показаний и выполняемой процедуры.
В целом, рецидив заболевания происходит у
40-75% реципиентов аутогенных ГСК трансплантатов
10-40% реципиентов аллогенных ГСК трансплантатов
В целом, показатели успешного лечения (без рака костного мозга) составляют
30-40% пациентов с рецидивом лимфомы, чувствительной к химиотерапии
20-50% пациентов с острым лейкозом в стадии ремиссии
По сравнению с применением только химиотерапии, трансплантация ГСК улучшает выживаемость больных с множественной миеломой. Показатель успешного лечения ниже у пациентов с более запущенным заболеванием или с реактивным солидным раком (например, опухоли эмбриональных клеток). Частота рецидивирования снижается у пациентов с болезнью трансплантат против хозяина (БТПХ), но в целом смертность увеличивается, если БТПХ протекает тяжело.
Интенсивный режим кондиционирования, эффективная профилактика БТПХ, курс лечения на основе циклоспорина и качественная поддерживающая терапия (например, при необходимости, антибиотиками, профилактика инфицирования вирусом герпеса и ЦМВ) увеличивают длительную выживаемость после ТГСК без рецидива заболевания.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Аллогенная трансплантация гемопоэтических стволовых клеток у детей с синдромом Гурлер
Для цитирования: Боровкова А. С., Киргизов К. И., Скоробогатова Е. В., Зубаровская Л. С. и др. Аллогенная трансплантация гемопоэтических стволовых клеток у детей с синдромом Гурлер // Доктор.Ру. Гематология. 2016. № 5 (122). С. 40–44.
Цель исследования: сравнить эффективность миелоаблативного кондиционирования (МАК) и кондиционирования редуцированной интенсивности (РИК) при аллогенной трансплантации гемопоэтических стволовых клеток (алло-ТГСК) у детей с синдромом Гурлер.
Материалы и методы. Проанализированы результаты алло-ТГСК, выполненной 31 пациенту с синдромом Гурлер (общее число трансплантаций — 33). Медиана возраста пациентов на момент установления диагноза составляла 15 месяцев (3–24), на момент аллогенной трансплантации — 22 месяца (9–42). В качестве подготовки к алло-ТГСК использовали МАК (24 трансплантации у 23 детей) или РИК (9 трансплантаций у 8 детей). Источники трансплантата — родственные и неродственные доноры.
Результаты. При медиане наблюдения около 50 месяцев (7–140) живы 26 пациентов. Четырехлетняя общая выживаемость составила 69,5 ± 0,16% независимо от интенсивности режима подготовки.
Заключение. При подготовке детей с синдромом Гурлер к алло-ТГСК эффективность режимов кондиционирования со сниженной и полной интенсивностью сопоставима.
Мукополисахаридоз I типа — лизосомная болезнь накопления с аутосомно-рецессивным типом наследования, обусловленная дефицитом фермента альфа-L-идуронидазы, ответственного за деградацию гликозаминогликанов. Самая тяжелая клиническая форма заболевания — синдром Гурлер. Заболеваемость синдромом Гурлер составляет 0,61–1,30 : 100 000 живорожденных [21].
В основе патогенеза синдрома Гурлер лежит накопление в тканях дерматансульфата и гепарансульфата, что приводит к поражению внутренних органов: ЦНС, сердечно-сосудистой и дыхательной систем, печени и селезенки, — а также к прогрессирующей задержке психомоторного развития, ухудшению зрения и слуха, органомегалии и деформации скелета [22].
Медиана возраста пациентов на момент установления диагноза составляет 10–12 месяцев [3, 15]. Ожидаемая продолжительность жизни больных синдромом Гурлер без лечения не превышает 10 лет (медиана — 5,3 года) [4, 21]. При своевременном начале ферментозаместительной терапии рекомбинантным препаратом ларонидазой улучшаются функции дыхательной и сердечно-сосудистой систем, повышается качество жизни пациентов, однако препарат, к сожалению, не проникает через гематоэнцефалический барьер и не предотвращает поражение ЦНС [11].
В настоящее время единственным радикальным методом лечения синдрома Гурлер служит аллогенная трансплантация гемопоэтических стволовых клеток (алло-ТГСК). Первая успешная алло-ТГСК при синдроме Гурлер выполнена в 1980 г. [10]. В настоящее время в мире накоплен большой положительный опыт лечения синдрома Гурлер с помощью алло-ТГСК; по данным разных авторов, долгосрочная выживаемость составляет 50–85% [1, 5, 8, 13, 14].
При выполнении алло-ТГСК у детей с болезнями накопления, в частности с синдромом Гурлер, традиционно применяют миелоаблативные режимы кондиционирования (МАК). Тем не менее эти режимы подготовки могут увеличивать риск развития тяжелых осложнений, приводя к повышению летальности, связанной с трансплантацией, особенно у пациентов с низким общесоматическим статусом, характерным для детей с синдромом Гурлер в продвинутой стадии заболевания (т. е. более старшего возраста). Показано, что летальность реципиентов алло-ТГСК с общесоматическим статусом менее 80% после трансплантации составляет 28,3% против 16% в группе детей, имевших статус Карновского в модификации Ланского 80–100% [17].
Вследствие поражения миокарда и коронарных сосудов мукополисахаридами у пациентов с синдромом Гурлер чаще развиваются фатальные сердечно-сосудистые осложнения [16], ввиду специфического поражения повышен риск развития веноокклюзионной болезни печени [20]. В отдаленном периоде после алло-ТГСК описаны нарастание неврологического дефицита и нарушения поведения, связанные и с токсичностью МАК.
Внедрение режимов кондиционирования сниженной интенсивности доз (кондиционирование редуцированной интенсивности — РИК) и повышение качества сопроводительной терапии позволили снизить токсичность подготовки пациентов с синдромом Гурлер к алло-ТГСК. Это особенно актуально для детей старшего возраста, у которых, как правило, более выражены клинические проявления основного заболевания [12]. С другой стороны, по данным различных исследований, применение РИК у пациентов с синдромом Гурлер увеличивает вероятность отторжения трансплантата и, как следствие, ухудшает общую выживаемость.
Цель работы: сравнить эффективность и токсичность МАК и РИК при подготовке к алло-ТГСК у детей с синдромом Гурлер.
МАТЕРИАЛЫ И МЕТОДЫ
Проанализированы результаты 33 алло-ТГСК, выполненных 31 пациенту с синдромом Гурлер в период с 2004 по 2015 г. в трех клиниках Российской Федерации: ФГБУ «Российская детская клиническая больница» МЗ РФ (главный врач — профессор, д. м. н. Н. Н. Ваганов), ФГБУ «Федеральный научно-клинический центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» МЗ РФ (генеральный директор — академик РАН, д. м. н., профессор А. Г. Румянцев), НИИ детской онкологии, гематологии и трансплантологии им. Р. М. Горбачевой Первого Санкт-Петербургского государственного медицинского университета им. И. П. Павлова МЗ РФ (директор — профессор, д. м. н. Б. В. Афанасьев).
Диагноз синдрома Гурлер устанавливали на основании клинических проявлений заболевания, результатов оценки активности альфа-L-идуронидазы, содержания гликозаминогликанов в моче и генетического исследования.
Анализ общей выживаемости выполнен методом Каплана — Мейера с использованием программных пакетов SPSS 17.0 и Statistica 8.0. Для вычисления медиан применяли табличный процессор Microsoft Office Excel 2007.
Медиана возраста на момент установления диагноза составила 15 месяцев (3–24), на момент алло-ТГСК — 22 месяца (9–42); РИК — 28 месяцев, МАК — 18 месяцев. В качестве подготовки к алло-ТГСК использовали МАК (24 трансплантации у 23 детей) или РИК (9 трансплантаций у 8 детей). В группе с РИК алло-ТГСК выполняли от неродственного донора, полностью совместимого по генам HLA-cистемы (10/10, n = 6) или имеющего различие с реципиентом в 1 гене (9/10, n = 3). В группе с МАК неродственная алло-ТГСК проведена 18 пациентам (10/10, n = 17; 9/10, n = 1), родственная — 5 пациентам.
В большинстве случаев в качестве источника гемопоэтических стволовых клеток (ГСК) использовали костный мозг (63,6%, n = 21), для 10 трансплантаций (30,3%) применяли периферические стволовые клетки крови (ПСКК), в 2 случаях (6,1%) — клетки пуповинной крови.
При РИК применяли мелфалан (в дозе 140 мг/м 2 ), флударабин (в дозе 150 мг/м 2 ) и антитимоцитарный иммуноглобулин (Атгам по 60 мг/кг). В основе МАК был треосульфан (42 г/м 2 ) в комбинации с флударабином, мелфаланом и антитимоцитарным иммуноглобулином (в случае алло-ТГСК от неродственного донора), в 4 случаях вместо треосульфана использовали бусульфан (в дозе 6 мг/кг), в 4 случаях мелфалан заменяли на тиотепу (10 мг/кг). Для профилактики острой реакции «трансплантат против хозяина» (РТПХ) применяли циклоспорин или такролимус в комбинации с метотрексатом или микофенолата мофетилом. Шести пациентам в группе с МАК перед алло-ТГСК дополнительно вводили ритуксимаб в дозе 375 мг/м 2 . Ввиду высокого содержания Т-лимфоцитов в трансплантате ПСКК у 3 пациентов в группе с РИК дополнительно проводили иммуномагнитную CD3/CD19-деплецию на приборе CliniMACS.
Восстановление гемопоэза фиксировали при достижении числа лейкоцитов в периферической крови выше 1000/мкл, нейтрофилов — более 500/мкл при отсутствии стимуляции гранулоцитарным колониестимулирующим фактором (Г-КСФ). Оценку донорского химеризма в периферической крови проводили на 30, 60, 90–100, 180 и 360-й дни после алло-ТГСК. Более 95% донорских клеток расценивали как полный донорский химеризм. В периоде после алло-ТГСК всем пациентам определяли активность альфа-L-идуронидазы. Клиническую стадию острой РТПХ и степень органного поражения оценивали в соответствии с общепринятой классификацией [18]. Выраженность клинических проявлений хронической РТПХ определяли по шкале NIH 2005 (NIH Consensus Conference, пересмотр 2014 г.), токсичность — по шкале NCI СТС ver. 3.0 (2006).
На момент анализа данных живы 26 пациентов с медианой наблюдения 50 месяцев (7–140).
У всех пациентов зафиксировано приживление трансплантата с достижением полного донорского химеризма к 30-му дню после ТГСК и нормальной активностью альфа-L-идуронидазы в лейкоцитах. Медиана восстановления числа нейтрофилов (более 500/мкл) составила 20 дней (11–29).
У 2 пациентов после РИК и 5 пациентов после МАК в различные сроки после алло-ТГСК зафиксирован смешанный гемопоэтический химеризм, с целью коррекции которого пациенты получали инфузии донорских лимфоцитов. В настоящее время у 2 из них сохраняется стабильный смешанный химеризм, у 5 пациентов (4 после МАК, 1 после РИК) отмечено вторичное отторжение трансплантата.
Четырехлетняя общая выживаемость после алло-ТГСК в общей группе составила 69,5 ± 0,16%. При сравнении общей выживаемости после использования МАК и РИК статистически значимых различий не получено: 72,75% и 75,00% соответственно (p = 0,24, рис. ).
Рис. Общая выживаемость пациентов с синдромом Гурлер после аллогенной трансплантации гемопоэтических стволовых клеток в зависимости от режима кондиционирования
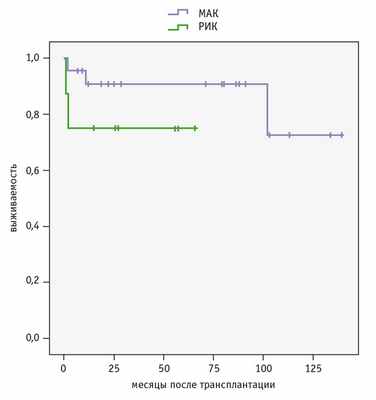
Осложнения у пациентов с синдромом Гурлер после аллогенной трансплантации гемопоэтических стволовых клеток в зависимости от режима кондиционирования
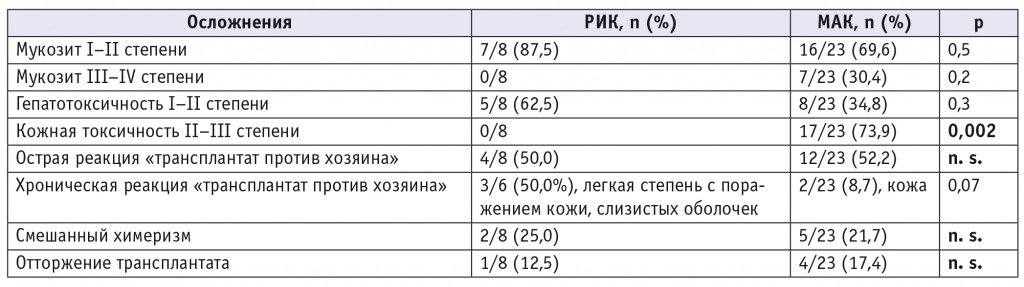
В группе пациентов, получивших МАК, во всех 3 случаях причиной смерти были инфекционные осложнения. Среди причин смерти после алло-ТГСК с РИК — острая РТПХ IV cтепени (1 пациент), TRALI-синдром (1 пациент).
Алло-ТГСК — единственный радикальный метод лечения, позволяющий значительно уменьшить выраженность клинических проявлений синдрома Гурлер, особенно поражения ЦНС.
Согласно большинству исследований, для больных синдромом Гурлер рекомендовано использование МАК [5, 19]. Однако у пациентов старшей возрастной группы с низким общесоматическим статусом применение МАК может быть ограничено. По данным Европейской группы по трансплантации крови и костного мозга (European Group for Blood and Bone Marrow Transplantation), оптимальным сроком выполнения алло-ТГСК является возраст до 2 лет, когда меньше степень мультиорганного вовлечения и возможно максимальное сохранение функций ЦНС. По литературным сведениям, у пациентов, перенесших трансплантацию в возрасте до 17 месяцев, зафиксирована лучшая бессобытийная выживаемость: 71% при трансплантации до 17 месяцев против 55% — после этого возраста [5].
Одной из причин выбора РИК в нашем исследовании послужил старший возраст пациентов, медиана которого в этой группе составила 28 месяцев. В одном мультицентровом исследовании медиана возраста — 13,5 месяца [2], по другим данным — 18 месяцев [9].
Частота развития смешанного химеризма и отторжения трансплантата у пациентов с синдромом Гурлер после алло-ТГСК с использованием МАК и РИК сопоставима (21,7% против 25,0% и 17,4% против 12,5%, p = n. s.), что не соответствует опубликованным сведениям об увеличении вероятности отторжения трансплантата при применении РИК у пациентов с синдромом Гурлер [1, 5 – 7].
При аллогенной трансплантации гемопоэтических стволовых клеток (алло-ТГСК) у детей с синдромом Гурлер эффективность режимов кондиционирования со сниженной интенсивностью доз (РИК) и с миелоаблативным кондиционированием (МАК) сопоставима. Режим кондиционирования следует окончательно выбирать с учетом состояния пациента, его возраста и коморбидного статуса. С целью выработки конкретных рекомендаций при использовании РИК и МАК необходимо дальнейшее наблюдение для оценки отдаленных осложнений алло-ТГСК и качества жизни пациентов.
Результаты применения кондиционирования с треосульфаном перед трансплантацией гемопоэтических стволовых клеток у пациентов с анемией Даймонда– Блекфена
Ключевые слова
Об авторах
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
Радыгина Светлана Анатольевна, врач-гематолог отделения
трансплантации гемопоэтических стволовых клеток №2
117997, Москва, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
117997, Москва, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
117997, Москва, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
117997, Москва, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
117997, Москва, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
117997, Москва, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
117997, Москва, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
117997, Москва, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
117997, Москва, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
117997, Москва, ул. Саморы Машела, 1
Список литературы
1. Vlachos A., Ball S., Dahl N., Alter B.P., Sheth S., Ramenghi U., et al.; Participants of Sixth Annual Daniella Maria Arturi International Consensus Conference. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol 2008; 142 (6): 859–76. DOI: 10.1111/j.1365-2141.2008.07269.x. Epub 2008 Jul 30. PMID: 18671700; PMCID: PMC2654478.
2. Josephs H.W. Anaemia of infancy and early childhood. Medicine 1936; 15: 307–451.
3. Diamond L.K., Blackfan K.D. Hypoplastic anemia. Am J Dis Child 1938; 56: 464–7.
4. Léger-Silvestre I., Caffey J.M., Dawaliby R., Alvarez-Arias D.A., Gas N., Bertolone S.J., et al. Specifi Role for Yeast Homologs of the Diamond Blackfan Anemia-associated Rps19 Protein in Ribosome Synthesis. J Biol Chem 2005; 280 (46): 38177–85. DOI: 10.1074/jbc.M506916200. Epub 2005 Sep 12. PMID: 16159874.
5. Lipton J.M., Ellis S.R. Diamond Blackfan anemia 2008–2009: broadening the scope of ribosome biogenesis disorders. Curr Opin Pediatr 2010; 22 (1): 12–9. DOI: 10.1097/MOP.0b013e328334573b. PMID: 19915471; PMCID: PMC2886588.
6. Da Costa L., O'Donohue M.F., van Dooijeweert B., Albrecht K., Unal S., Ramenghi U., et al. Molecular approaches to diagnose Diamond–Blackfan anemia: The EuroDBA experience. Eur J Med Genet 2018; 61 (11): 664–73. DOI: 10.1016/j.ejmg.2017.10.017. Epub 2017 Oct 26. PMID: 29081386.
7. Bartels M., Bierings M. How I manage children with Diamond–Blackfan anaemia. Br J Haematol 2019; 184 (2): 123–33. DOI:10.1111/bjh.15701. Epub 2018 Dec 4. PMID: 30515771; PMCID: PMC6587714.
8. Ruggero D., Pandolf P.P. Does the ribosome translate cancer? Nat Rev Cancer 2003; 3 (3): 179–92. DOI: 10.1038/nrc1015.PMID: 12612653.
10. Lipton J.M., Atsidaftos E., Zyskind I., Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer 2006; 46 (5):558–64. DOI: 10.1002/pbc.20642. PMID:16317735.
11. Berdoukas V., Nord A., Carson S., Puliyel M., Hofstra T., Wood J., et al. Tissue iron evaluation in chronically transfused children shows signifiant levels of iron loading at a very young age. Am J Hematol 2013; 88 (11): E283–5. DOI: 10.1002/ajh.23543.Epub 2013 Sep 2. PMID: 23861216.
12. Bartels M., Bierings M. How I manage children with Diamond–Blackfan anaemia. Br J Haematol 2019; 184 (2): 123–33. DOI:10.1111/bjh.15701. Epub 2018 Dec 4. PMID: 30515771; PMCID: PMC6587714.
13. Peffult de Latour R., Peters C., Gibson B., Strahm B., Lankester A., de Heredia C.D.; Pediatric Working Party of the European Group for Blood and Marrow Transplantation; Severe Aplastic Anemia Working Party of the European Group for Blood and Marrow Transplantation. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant 2015; 50 (9): 1168–72. DOI: 10.1038/bmt.2015.117.Epub 2015 Jun 8. PMID: 26052913.
15. Pospisilova D., Cmejlova J., Hak J., Adam T., Cmejla R. Successful treatment of a Diamond-Blackfan anemia patient with amino acid leucine. Haematologica 2007; 92 (5): e66–7. DOI: 10.3324/haematol.11498. PMID: 17562599.
16. Dalle J.H., Peffult de Latour R. Allogeneic hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Int J Hematol 2016; 103 (4):373–9. DOI: 10.1007/s12185-016-1951-0.Epub 2016 Feb 12. PMID: 26872907.
17. Alter B.P. Inherited bone marrow failure syndromes: considerations pre- and posttransplant. Hematology Am Soc Hematol Educ Program 2017; 2017 (1): 88–95.
19. Fagioli F., Quarello P., Zecca M., Lanino E., Corti P., Favre C., et al. Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: A report from the Italian Association of Paediatric Haematology and Oncology Registry. Br J Haematol 2014; 165 (5): 673–81.
20. Vlachos A., Federman N., ReyesHaley C., Abramson J., Lipton J.M. Hematopoietic stem cell transplantation for Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Bone Marrow Transplant 2001; 27 (4): 381–6.
21. Strahm B., Loewecke F., Niemeyer C.M., Albert M., Ansari M., Bader P. Favorable outcomes of hematopoietic stem cell transplantation in children and adolescents with Diamond-Blackfan anemia. Blood Adv 2020; 4 (8): 1760–9. DOI: 10.1182/bloodadvances.2019001210. PMID: 32343795; PMCID: PMC7189291.
22. Przepiorka D., Weisdorf D., Martin P., Klingemann H.G., Beatty P., Hows J., Thomas E.D. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant 1995; 15 (6): 825–8. PMID:7581076.
23. Burroughs L.M., Shimamura A., Talano J.A., Domm J.A., Baker K.K., Delaney C., et al. Allogeneic Hematopoietic Cell Transplantation Using Treosulfan-Based Conditioning for Treatment of Marrow Failure Disorders. Biol Blood Marrow Transplant 2017; 23 (10): 1669–77. DOI: 10.1016/j.bbmt.2017.06.002. Epub 2017 Jun 7. PMID: 28602958; PMCID: PMC5605451.
24. Bartelink I.H., Bredius R.G., Belitser S.V., Suttorp M.M., Bierings M., Knibbe C.A., et al. Association between busulfan exposure and outcome in children receiving intravenous busulfan before hematologic stem cell transplantation. Biol Blood Marrow Transplant 2009; 15 (2): 231–41. DOI: 10.1016/j.bbmt.2008.11.022. PMID:19167683.
25. Balashov D., Shcherbina A., Maschan M., Trakhtman P., Skvortsova Y., Shelikhova L., et al. Single-Center Experience of Unrelated and Haploidentical Stem Cell Transplantation with TCRab and CD19 Depletion in Children with Primary Immunodefiiency Syndromes. Biol Blood Marrow Transplant 2015; 21: 1955–62.
26. Maschan M., Shelikhova L., Ilushina M., Kurnikova E., Boyakova E., Balashov D., et al. TCR-alpha/beta and CD19 depletion and treosulfan-based conditioning regimen in unrelated and haploidentical transplantation in children with acute myeloid leukemia. Bone Marrow Transplant 2016; 51 (5): 668–74. DOI: 10.1038/bmt.2015.343.Epub 2016 Jan 25.
Клинические особенности кожной формы острой реакции «трансплантат против хозяина» при аллогенной трансплантации гемопоэтических стволовых клеток у детей с онкогематологическими заболеваниями
Реакция «трансплантат против хозяина» (РТПХ) — частое осложнение, возникает у половины больных после аллогенной трансплантации гемопоэтических стволовых клеток (ТГСК), является одной из основных причин смертности пациентов, не связанной с рецидивом основного заболевания. Поражение кожи в симптомокомплексе острой РТПХ развивается в течение первых 100 дней после ТГСК, представляет собой сложную диагностическую и терапевтическую проблему. Выраженный иммуносупрессивный статус детей в раннем посттрансплантационном периоде усиливает и видоизменяет течение дерматозов, инфекционных поражений, проявлений медикаментозной токсичности, приводя к запуску иммуноаллергических процессов с возможностью трансформации в генерализованные жизнеугрожающие поражения кожи и слизистых оболочек. В то же время токсические, аллергические, инфекционные поражения кожи могут присутствовать в клинической картине одномоментно или развиваться последовательно. В связи с относительно небольшой частотой проводимых ТГСК у детей с онкологическими заболеваниями описание клинической картины поражений кожи при острой РТПХ представляет несомненный интерес и научно-практическую значимость. В статье обобщены данные об этиологии, патогенезе, клинических формах, методах диагностики и лечения кожных осложнений раннего посттрансплантационного периода при ТГСК.
Ключевые слова
Об авторах
Белышева Татьяна Сергеевна, доктор медицинских наук, ведущий научный сотрудник научно-консультативного отделения НИИ детской онкологии и гематологии
115478, Москва, Каширское ш., д. 24
Остальные авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить
Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина
Россия
Москва
Раскрытие интересов:
Остальные авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить
Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина
Россия
Москва
Раскрытие интересов:
Остальные авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить
Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина
Россия
Москва
Раскрытие интересов:
Остальные авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить
Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина
Россия
Москва
Раскрытие интересов:
Остальные авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить
Национальный медицинский исследовательский центр здоровья детей; Первый Московский государственный медицинский университет им. И.М. Сеченова (Сеченовский Университет); Центральная государственная медицинская академия Управления делами Президента РФ; НИИ педиатрии и охраны здоровья детей ЦКБ РАН
Россия
Москва
Раскрытие интересов:
Н. Н. Мурашкин — получение исследовательских грантов от фармацевтических компаний Jansen, Eli Lilly, Novartis. Получение гонораров за научное консультирование от компаний Galderma, Pierre Fabre, Bayer, LEO Pharma, Pfizer, AbbVie, Amryt Pharma
Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина
Россия
Москва
Раскрытие интересов:
Остальные авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить
Остальные авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить
Список литературы
2. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005;11(12):945–956. doi: 10.1016/j.bbmt.2005.09.004.
3. Sung AD, Chao NJ. Acute graft-versus-host disease: are we close to bringing the bench to the bedside? Best Pract Res Clin Haematol. 2013;26(3):285–292. doi: 10.1016/j.beha.2013.10.009.
5. Paun O, Phillips T, Fu P, et al. Cutaneous complications in hematopoietic cell transplant recipients: impact of biopsy on patient management. Biol Blood Marrow Transplant. 2013;19(8): 1204–1209. doi: 10.1016/j.bbmt.2013.05.006.
7. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant. 2015; 21(3):389–401.e1. doi: 10.1016/j.bbmt.2014.12.001.
8. Pidala J, Vogelsang G, Martin P, et al. Overlap subtype of chronic graft-versus-host disease is associated with an adverse prognosis, functional impairment, and inferior patient-reported outcomes: a Chronic Graft-versus-Host Disease Consortium study. Haematologica. 2012;97(3):451–458. doi: 10.3324/haematol.2011.055186.
9. Saliba RM, de Lima M, Giralt S, et al. Hyperacute GVHD: risk factors, outcomes, and clinical implications. Blood. 2007;109(7): 2751–2758. doi: 10.1182/blood-2006-07-034348.
10. Zeiser R, Penack O, Holler E, Idzko M. Danger signals activating innate immunity in graft-versus-host disease. J Mol Med (Berl). 2011;89(9):833–845. doi: 10.1007/s00109-011-0767-x.
11. Harris AC, Young R, Devine S, et al. International, multi-center standardization of acute graft-versus-host disease clinical data collection: a report from the MAGIC consortium. Biol Blood Marrow Transplant. 2016;22(1):4–10. doi: 10.1016/j.bbmt.2015.09.001.
12. Ion D, Stevenson K, Woo SB, et al. Characterization of oral involvement in acute graft-versus-host disease. Biol Blood Marrow Transplant. 2014;20(11):1717–1721. doi: 10.1016/j.bbmt.2014.06.031.
13. Surjana D, Robertson I, Kennedy G, et al. Acute cutaneous graft-versus-host disease resembling type II (atypical adult) pityriasis rubra pilaris. Australas J Dermatol. 2015;56(1):e21–e23. doi: 10.1111/ajd.12108.
14. Huang J, Pol-Rodriguez M, Silvers D, Garzon MC. Acquired ichthyosis as a manifestation of acute cutaneous graft-versushost disease. Pediatr Dermatol. 2007;24(1):49–52. doi: 10.1111/j.1525-1470.2007.00333.x.
15. Matsushita T, Hasegawa M, Shirasaki F, et al. A case of acute cutaneous graft-versus-host disease mimicking psoriasis vulgaris. Dermatology. 2008;216(1):64–67. doi: 10.1159/000109361.
16. Betts BC, Young JA, Ustun C, et al. Human herpesvirus 6 infection after hematopoietic cell transplantation: is routine surveillance necessary? Biol Blood Marrow Transplant. 2011;17:1562–1568. doi: 10.1016/j.bbmt.2011.04.004.
17. Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59(3): 524–529. doi: 10.1016/j.jaad.2008.05.018.
18. Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011; 24(4):411–423. doi: 10.1111/j.1529-8019.2011.01436.x.
20. Horn TD, Beveridge RA, Egorin MJ, et al. Observations and proposed mechanism of N,N’,N’’-triethylenethiophosphoramide (thiotepa)-induced hyperpigmentation. Arch Dermatol. 1989; 125:524–527.
21. Callen J, Jorizzo J, Bolognia J, et al. Dermatological Signs of Internal Disease. 4th Ed. Saunders; 2009. р. 323.
22. Mays SR, Kunishige JH, Truong E, et al. Approach to the morbilliform eruption in the hematopoietic transplant patient. Semin Cutan Med Surg. 2007;26(3):155–162. doi: 10.1016/j.sder.2007.09.004.
23. Peñas PF, Zaman S. Many faces of graft-versus-host disease. Australas J Dermatol. 2010;51(1):1–10. doi: 10.1111/j.1440-0960.2009.00577.x.
24. Byun HJ, Yang JI, Kim BK, Cho KH. Clinical differentiation of acute cutaneous graft-versus-host disease from drug hypersensitivity reactions. J Am Acad Dermatol. 2011;65(4):726–732. doi: 10.1016/j.jaad.2010.07.042.
25. Hillen U, Hausermann P, Massi D, et al. Consensus on per forming skin biopsies, laboratory workup, evaluation of tissue samples and reporting of the results in patients with suspected cutaneous graft-versus-host disease. J Eur Acad Dermatol Venereol. 2015; 29(5):948–954. doi: 10.1111/jdv.12737.
26. Penack O, Marchetti M, Ruutu T, et al. Prophylaxis and management of graft versus host disease after stem-cell transplantation for haematological malignancies: updated consensus recommendations of the European Society for Blood and Marrow Transplantation. Lancet Haematol. 2020;7(2):e157–e167. doi: 10.1016/S2352-3026(19)30256-X.
27. Manalo IF, Miller IA, Davies LS. More immune dysregulation: sarcoidosis and chronic graft-versus-host disease after allogeneic stem cell transplant. JAAD Case Rep. 2016;2(2):138–140. doi: 10.1016/j.jdcr.2016.01.008.
28. Paczesny S, Braun TM, Levine JE, et al. Elafin is a biomarker of graft versus host disease of the skin. Sci Transl Med. 2010;2(13): 1–19. doi: 10.1126/scitranslmed.3000406.
29. Levine JE, Braun TM, Harris AC, et al. A prognostic score for acute graft-versus-host disease based on biomarkers: a multicentre study. Lancet Haematol. 2015;2(1):21–29. doi: 10.1016/S2352-3026(14)00035-0.
30. Pidala J, Sigdel TK, Wang A, et al. A combined biomarker and clinical panel for chronic graft versus host disease diagnosis. J Pathol Clin Res. 2017;3(1):3–16. doi: 10.1002/cjp2.58.
Читайте также: