Ювенильная апоневротическая фиброма. Фиброзная гамартома младенцев.
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
Частота случаев заболеваний ежегодно среди детей европеоидной популяции в возрасте до 15 лет составляет 8,4:1 млн, а среди детей негроидной популяции — около 4,2: 1 млн. Из всех видов сарком мягких тканей на рабдомиосаркому приходится около 50% (см. табл 19-11).
Подтипы сарком мягких тканей могут быть идентифицированы по сходству с интактными тканями, из которых они исходят (табл. 19-11). Некоторые из них отличаются агрессивным ростом, другие растут относительно медленно. У большинства больных саркома представляет собой единое образование.
Таблица 19-11. Саркомы мягких тканей
Естественное течение процесса
Первичная мезенхима
Соединительные
ткани
Жировая
Злокачественная
мезенхимома
Липосаркома
Может быть врожденной, обычно локализуется в конечностях или забрюшинном пространстве; характеризуется быстрым ростом и часто рецидивирует; метастазирует обычно в легкие, мозг, печень и лимфатические узлы Редко встречается у детей, развивается в основном в предсуществовавшей липоме, отличается быстрым ростом, иногда сопровождается общей симптоматикой (лихорадочное состояние); метастазы, относительно частые у
Ткань, из которой исходит опухоль
Естественное течение процесса
взрослых, реже встречаются у детей; исход зависит от степени дифференциации клеток Может быть врожденной; больные в возрасте до 5 лет (т. е. при инфантильной форме) отличаются большей выживаемостью, чем в возрасте старше 15 лет (метастазы в 7,5% и 50% соответственно); у лиц в возрасте после 15 лег местный рецидив в 40% чаще всего локализуется в конечностях; первичный очаг обычно связан с предшествующим облучением
Медленно растущая опухоль, чаще встречающаяся на туловище и волосистой части головы или лица; прогрессирует от кожного узелка до выраженной опу холи на ножке в течение нескольких лет, но иногда (редко) растет быстро; высока частота местных рецидивов; распространяется на подкожные ткани, мышцы и кости; метастазирует главным образом в легкие и мозг
Фиброматозы (фиброзная гамартома, febrosis colli, инфантильная и ювенильная апоневротическая фиброма, врожденный генерализованный фиброматоз, инфантильная фиброма пальцев)
Отличается инвазивным ростом, редко метастазирует (исключение составляет врожденный генерализованный фибро матоз обычно внутренних органов, имеющий неблагоприятный прогноз); прогноз в основном благоприятный, но частота рецидивов составляет 90%
Злокачественный
фиброзный
гистиоцитоз
Редко встречается в возрасте до 40 лет; относится к виду сарком мягких тканей, которыми заболевают во вторую половину жизни; типично расположение на конечностях (особенно на бедре) и в забрюшинном пространстве, исходит из глубоких фасций или мышц; местные рецидивы в 44% случаев; прогноз зависит от размера и первичной глубины опухоли (т. е. поражена ли только фасция или более глубокие структуры)
Медленно растущая и инфильтрирующая опухоль; обычно проявляется болезненными узелками на конечностях (особенно на предплечьях или кисти, в под-
Ткань, из которой исходит опухоль
Естественное течение процесса
коленной области); узелки часто покрыты изъязвленной кожей; высокая частота местных рецидивов; часто метастазирует в легкие, лимфатические узлы и кожу
Встречается очень редко, локализуется в основном на конечностях, иногда появляется через несколько десятилетий после врожденной или приобретенной лимфедемы; быстро прогрессирует с метастазами в грудную стенку и плевру
Быстро прогрессирует с высокой частотой летальных исходов; в первую очередь поражает конечности, печень и область головы и шеи; связана с введением торотраста и винилхлорида; может развиваться у детей в возрасте 1 года; метастазирует в печень, кости и надпочечники
Может проявляться как опухоль; наиболее частая первичная локализация — на конечностях и туловище; высокая частота местных рецидивов (50%); метастазы появляются поздно (через 10 лет), обычно в легкие, мозг и печень
Первичная локализация в носоглотке; не метастазирует, но растет инвазивно
Встречается редко и главным образом у лиц в возрасте 20—40 лет; в 80—90% случаев в области коленного сустава, стопы и кисти; обычно метастазирует в легкие, кости и лимфатические узлы; характеризуется местными рецидивами с поздними проявлениями (даже через 10—15 лет)
Альвеолярная саркома мягких тканей
Медленно растущая опухоль с первичной локализацией в основном на бедре или передней стенке живота; часты местные рецидивы и метастазы, главным образом поздние и чаще в легкие, кости и мозг; встречается у детей в возрасте 3 лет
Мезотелиаль-
ная
Мышечная ткань
Редко встречается у детей; обусловлена контактом с асбестом у взрослых; первичная локализация чаще в плевре и брюшине; быстро прогрессирует с широким местным распространением и метастазами
Редко встречается у детей; средний возраст заболевших составляет 23 года; чаще начинается на передней брюшной стенке или в плечевой борозде; проявляется в основном в виде фиксиро-
Ткань, из которой исходит опухоль
Естественное течение процесса
ванного болезненного образования; может зависеть от уровня эстрогенов и развивается в послеродовой период'
Редко встречается у детей, описаны случаи заболевания новорожденных; чаще локализуется в желудочно-кишечном тракте, мочеполовой и дыхательной системах; метастазирует в основном в печень, регионарные лимфатические узлы, легкие, брюшину и поджелудочную железу
В модификации из: Rabdomyosarcoma and other soft tissue sarcomas/Ed. A. S. Levine.— Cancer in the Young.—New York: Masson Publishing, 1982, pp. 615—632.
В этом случае важно определить ее распространенность до хирургического вмешательства, особенно в отношении метастазов в кости и легкие. Иногда радикальная операция служит единственным шансом излечения. Вероятно, как ни при одной другой группе опухолей, в этих случаях необходимо тщательно исследовать опухолевую ткань для определения ее специфического типа и выявления злокачественной или доброкачественной природы. Послеоперационная химиотерапия проводится при злокачественных опухолях с выраженной митотической активностью независимо от их размера и возможности резекции.
Ювенильная апоневротическая фиброма. Фиброзная гамартома младенцев.
Ювенильная апоневротическая фиброма. Фиброзная гамартома младенцев.
Ювенильиая апоневротическая фиброма (хрящевой аналог фиброматоза, ювенильная кальцифицирующаяся фиброма) впервые описана L. E. Keasbey в 1953 г. Возникает в возрасте 3—15 лет, но может наблюдаться и у взрослых. Локализуется главным образом на кисти и стопе, чаще у лиц мужского пола. Поражение представляет нечетко отграниченный, часто связанный с апоневрозом, плотный узел сероватого цвета, хрустящий при разрезе.
Может инфильтрировать подлежащие ткани вплоть до кости. Диаметр его не превышает 3 см, хотя описаны и узлы диаметром 6—10 см. Микроскопически узел образован веретенообразными и овальными клетками с «пухлыми» овальными ядрами н светлой гомогенной цитоплазмой. Клетки располагаются то компактно, особенно на периферии, то рыхло. Характерной чертой строения являются фокусы кальцификации и хрящевой трансформации.
Процесс часто рецидивирует, особенно при локализации на кисти и стопе, что, вероятно, связано с трудностью иссечения Учитывая наклонность к рецидивам, большинство исследователей относят его к фиброматозам.
Фиброзная гамартома младенцев (синоним субэпидермальная фиброзная опухоль младенцев) впервые описана R. D. К. Reye в 1956 г, а под настоящим названием выделена F. M. Enzinger в 1965 г. Большинство исследователей склонны рассматривать этот процесс как вариант фиброматоза, а не как «гамартому». Она обнаруживается с момента рождения и до 4-летнего возраста, преимущественно в первые 2 года жизни; среди больных преобладают мальчики (до 70% наблюдений).
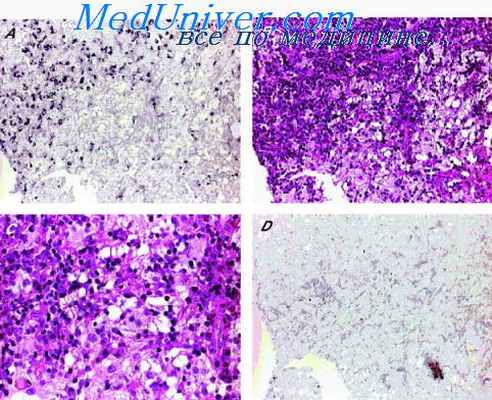
Образование располагается главным образом в подкожной клетчатке и локализуется преимущественно на предплечье, плече, в подмышечной и ягодичной областях. Могут наблюдаться множественные поражения. Очаг поражения представляет собой нечетко контурированный узел серовато-желтого цвета на разрезе, обычно диаметром не более 4 см, хотя описаны узлы размером 7—10 см.
Микроскопически узел характеризуется наличием 3 основных структурных компонентов: фиброзная ткань разной степени клеточности в виде тяжей и лучков, очаги, сходные с примитивной мезенхимой, зрелая жировая ткань, занимающая около половины объема узла. Некоторые авторы отмечают также значительное количество сосудов капиллярного типа Прогноз благоприятный, рецидивы после иссечения наблюдаются редко По данным А. М. Вихерта (1969), образование эволюционирует в сторону созревания всех тканевых компонентов и к 4 годам может превратиться в келоид с прослойками зрелой жировой ткани.
Ювенильная ангиофиброма носоглотки описана S. S. Sternberg в 1954 г. Встречается преимущественно у лиц мужского пола, главным образом между 10 и 20 годами. Клиническая симптоматика связана с величиной очага и степенью распространения его на соседние области. Происходит из carlilago basalis, состоит из соединительной ткани с умеренным количеством клеток, последние часто с уродливыми ядрами. В образовании содержатся довольно равномерно расположенные сосуды синусоидного типа. Характерна инвазия в соседние ткани, включая кости. Случаев спонтанной регрессии не наблюдали, возможна только частичная регрессия.
Наследственный гингивальный фиброматоз (наследственная гиперплазия десен, первичная генерализованная гипертрофия десен, врожденный идиопатический гингивальный фиброматоз, врожденная макрогингивия, гигантизм десен, множественные эпулиды) детально описан D Winstock (1964), Е. Р. Hehefer, L. А. Кау (1967), G. Farrer-Brown et al (1972). Встречается одинаково часто у лиц обоего пола и выражается опухолевидным утолщением десен с расшатыванием и выпадением зубов. Процесс может возникать с момента прорезывания молочных зубов, а иногда с рождения, чаще является наследственным.
Иногда сочетается с гипертрихозом, нейрофиброматозом, слабоумнем, херувизмом. Гистологически отмечается разрастание плотной аваскулярной соединительной тканн с малым количеством клеток и большим количеством коллагена, что придает процессу сходство с келоидом. Воспалительные изменения минимальны. Описан также клеточный и сосудистый вариант с крупными фибробластами, мелкими фокусами костеобразования и немногочисленными клетками типа остеокластов. Рецидивы возможны Спонтанной регрессии не описано.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Фиброзная гамартома младенцев редкой локализации, клинические случаи
Фиброзная гамартома младенцев (ФГМ) является редкой опухолью, составляющей менее 2 % опухолей мягких тканей, возникающих на первом году жизни ребенка. Опухоль возникает из подкожной клетчатки и наиболее часто обнаруживается в подмышечной впадине, затем в плечевой, паховой областях и грудной стенке. Характерным микроскопическим видом ФГМ является наличие 3 типов тканей в различных пропорциях: четко очерченные пучки плотной волокнистой соединительной ткани, примитивной мезенхимы (организованной в гнездах, концентрических завитках или полосах) и промежуточной зрелой жировой ткани. Хирургическое удаление является основным методом выбора лечения при данной патологии. Мы сообщаем о 2 случаях редкого расположения ФГМ, локализующихся в ягодичной области и области задней верхней трети бедра. При этом в одном наблюдении поражение затрагивало только подкожно-жировую клетчатку, а во втором отмечался распространенный инвазивный процесс в мышцах. Несмотря на доброкачественный характер опухоли, распространение ее характеризуется инвазивным ростом (не имеет капсулы и четкой границы с окружающими тканями), что может вызвать трудности у хирургов при первичной операции в определении границ удаления ФГМ. Считаем целесообразным использовать для этого экспресс-биопсию во время оперативного вмешательства.
Ключевые слова
Об авторах
Илья Маркович Каганцов, д.м.н., доцент, заведующий научно-исследовательской лабораторией хирургии врожденной и наследственной патологии, SPIN-код: 7936-8722, AuthorID: 333925
к.м.н., доцент, заведующая отделением детской хирургии пороков развития, SPIN-код: 6863-7436, AuthorID: 647729
врач-детский хирург отделения детской хирургии пороков развития
врач-детский хирург отделения детской хирургии пороков развития, SPIN-код: 2992-1533
к.м.н., врач-патологоанатом, старший научный сотрудник РНХИ им. проф. А.Л. Поленова – филиала, SPIN-код: 3327-7617, AuthorID: 1018053
к.м.н., заведующая научно-исследовательской лабораторией детской нейроиммуноонкологии Центра персонализированной медицины и заведующая отделением химиотерапии онкогематологических заболеваний и трансплантации костного мозга для детей, SPIN-код: 1776-6462, AuthorID: 808399
Список литературы
1. Reye R.D. A consideration of certain subdermal fibromatous tumours of infancy. J Pathol Bacteriol. 1956;72(1):149–54. doi: 10.1002/path.1700720120.
2. Enzinger F.M. Fibrous hamartoma of infancy. Cancer. 1965;18:241–8. doi: 10.1002/1097-0142(196502)18:23.0.co;2-c.
4. Al-Ibraheemi A., Martinez A., Weiss S.W., Kozakewich H.P., Perez-Atayde A.R., Tran H., Parham D.M., Sukov W.R., Fritchie K.J., Folpe A.L. Fibrous hamartoma of infancy: a clinicopathologic study of 145 cases, including 2 with sarcomatous features. Mod Pathol. 2017;30(4):474–85. doi: 10.1038/modpathol.2016.215.
5. Carretto E., DallʼIgna P., Alaggio R., Siracusa F., Granata C., Ferrari A., Cecchetto G. Fibrous hamartoma of infancy: an Italian multi-institutional experience. J Am Acad Dermatol. 2006;54(5):800–3. doi: 10.1016/j.jaad.2005.11.1076.
6. Efem S.E., Ekpo M.D. Clinicopathological features of untreated fibrous hamartoma of infancy. J Clin Pathol. 1993;46(6):522–4. doi: 10.1136/jcp.46.6.522.
7. Coffin C.M., Alaggio R. Fibroblastic and myofibroblastic tumors in children and adolescents. Pediatr Dev Pathol. 2012;15(1 Suppl):127–80. doi: 10.2350/10-12-0944-PB.1.
8. Dickey G.E., Sotelo-Avila C. Fibrous hamartoma of infancy: current review. Pediatr Dev Pathol. 1999;2(3):236–43. doi: 10.1007/s100249900119.
9. Stensby J.D., Conces M.R., Nacey N.C. Benign fibrous hamartoma of infancy: a case of MR imaging paralleling histologic fi ndings. Skeletal Radiol. 2014;43(11):1639–43. doi: 10.1007/s00256-014-1940-4.
10. Ji Y., Hu P., Zhang C., Yan Q., Cheng H., Han M., Huang Z., Wang X., Li H., Han Y. Fibrous hamartoma of infancy: radiologic features and literature review. BMC Musculoskelet Disord. 2019;3;20(1):356. doi: 10.1186/s12891-019-2743-5.
12. Imaji R., Goto T., Takahashi Y., Akiyama T., Yamadori I. A case of recurrent and synchronous fibrous hamartoma of infancy. Pediatr Surg Int. 2005;21(2):119–20. doi: 10.1007/s00383-004-1357-6.
13. Yano S., Hida K., Nagashima K., Iwasaki Y. Spinal fibrous hamartoma of infancy: case report. Neurosurgery. 2004;55(3):712. doi: 10.1227/01.neu.0000134614.73236.a4.
14. Arioni C., Bellini C., Oddone M., Risso F.M., Scopesi F., Nozza P., Serra G., Tomà P. Congenital fibrous hamartoma of the knee. Pediatr Radiol. 2006;36(5):453–5. doi: 10.1007/s00247-006-0126-1.
15. You M.H., Shin D.H., Choi J.S., Kim B.S., Kim Y., Kim J. The First Reported Case of Fibrous Hamartoma of Infancy with Hyperhidrosis and Hypertrichosis in Korea. J Korean Med Sci. 2018;33(9):e66. doi: 10.3346/jkms.2018.33.e66.
16. Stepančec H., Kokot Z., Keretić D., Radiković S., Grgurović D. Fibrous Hamartoma of Infancy in the Scrotum. European J Pediatr Surg Rep. 2019;7(1):e100–3. doi: 10.1055/s-0039-1697924.
17. Wang S., Ma Q., Ying H., Jiao Q., Yang D., Zhang B., Zhao L. Giant fibrous hamartoma of infancy: A case report. Medicine (Baltimore). 2020;99(11):e19489. doi: 10.1097/MD.0000000000019489.
18. Miroux-Catarino A., Claro C., Viana I. Giant fibrous hamartoma of infancy: pitfall of CD34 positive dermal mesenchymal tumor. Dermatol Online J. 2018;24(6):13030/qt160398hv. PMID: 30142714.
19. Sehgal M., Anand S., Dhua A.K., Yadav D.K., Arava S., Barwad A. Rare Paratesticular Masses in Children. J Indian Assoc Pediatr Surg. 2021;26(2):117–9. doi: 10.4103/jiaps.JIAPS_182_19.
20. Agrawal L., Bansal R., Singh J., Sharma S. Fibrous hamartoma of infancy in an unusual location – a case report. Gulf J Oncolog. 2010;(8):52–4. PMID: 20601341.
21. Шароев Т.А., Сухарев А.В., Иванова Н.М., Климчук О.В., Рощин В.Ю. Инфантильная фибросаркома мягких тканей бедра у ребенка 3 месяцев. Российский вестник детской хирургии, анестезиологии и реаниматологии. 2014;4(2):81–6.
Другие доброкачественные новообразования соединительной и других мягких тканей
Неметастазирующие опухолеподобные миофибро-, фибробластические процессы неизвестной этиологии.
Эпидемиология. Большинство фиброматозов встречаются только у детей.
Гистологическая классификация доброкачественных опухолей фиброзной ткани
■ Фиброзная гамартома младенцев.
■ Солитарный и мультицентрический фиброматозы.
■ Обызвествляющая апоневротическая фиброма (редко у детей).
■ Инфантильный дигитальный фиброматоз.
■ Десмоидный фиброматоз (редко у детей).
■ Неклассифицируемые фиброматозы (редко у детей).
Фиброзная гамартома младенцев
Эпидемиология. Обнаруживается у детей до 1-го года. В 20% врожденная. Чаще болеют мальчики.
Клиническая картина. Преимущественно одиночное образование с нечеткими границами, находится в коже и подкожно-жировой клетчатке.
Локализация. Подмышечные впадины, проксимальные отделы конечностей, паховая область.
Дифференциальный диагноз. C другими вариантами фиброматозов и инфантильной фибросаркомой.
Эпидемиология. Чаще поражаются мальчики.
Клиническая картина и локализация. Клинические признаки появляются на 2-4-й нед. жизни: развивается кривошея, на фоне которой выявляется новообразование в нижней (чаще) или в верхней (реже) частях кивателыюй мышцы, чаще справа. Стадия роста сменяется стабилизацией и последующей регрессией.
Дифференциальный диагноз. Кривошея.
Инфантильный миофиброматоз (синонимы: врожденный множественный фиброматоз, врожденный генерализованный фиброматоз, агрессивный инфантильный фиброматоз).
Эпидемиология. Чаще всего обнаруживается с рождения или в течение 1-го года жизни.
Значительно реже у детей старше 2 лет. Чаще у мальчиков. Солитарная форма — 75%, множественная — 25%.
Клиническая картина и локализация. Одиночные безболезненные узлы размерами 2-5 см в диаметре, могут располагаться в подкожножировой клетчатке, мышцах, редко — в костях и органах. При расположении во внутренних органах клинические проявления зависят от локализации, сдавления прилежащих нервных стволов; при поражении ЖКТ — гипотрофия, гемоколит.
Десмоидный фиброматоз (синонимы: инфантильный/ювенильный де- смоидоподобный фиброматоз, мышечно-апоневротический фиброматоз, агрессивный фиброматоз).
Неметастазирущее новообразование, локализующееся преимущественно в области головы, шеи, плечевого пояса и брюшной стенки. Имеет склонность к рецидивировапию. Характерен также инфильтрирующий рост вдоль фасциальных структур с местным и отдаленным распространением.
Эпидемиология. Встречается в 2 раза чаще у мальчиков, чем у девочек, в то время как у взрослых бывает значительно чаще у женщин. Средний возраст при постановке диагноза — 10 лет.
Клиническая картина и локализация. Опухолевидное безболезненное образование в типичном месте с медленным ростом.
Лечение. Широкое иссечение, иногда ампутация (при рецидивах).
Прогноз. Вероятность рецидива даже после адекватной операции очень высока. Поэтому целесообразно тщательное наблюдение с PKT- и МРТ-конт- ролем каждые 2-3 мес.
Фиброма. Эластофиброма. Фиброматоз.
Термин «фиброма» используют излишне широко н часто применяют к доброкачественным поражениям с выраженным коллагенообразованием и таким, как нейрофибромы, некоторые репаративные процессы и фиброматозы. Нередко в фиброме встречаются включения жировой ткани. В этих случаях опухоль называют фибролипомой R. Lattes (1982) считает, что одним из характерных типов фибромы является педункулярное (имеющее ножку) или нитевидное врожденное образование, сформированное нормальными фибробластическими элементами дермы и покрытых эпидермисом. Это безобидное, редко удаляемое, в основном по косметическим соображениям, новообразование.
Эластофиброма (спины) описана впервые О. Н. Jarv A. E. Saxen. Редко встречающаяся, нечетко отграниченная узловатая опухоль, локализующаяся преимущественно на спине с фасциями и мышцами под лопаткой н в межлопаточной области. Может достигать 10—15 см в поперечнике Возникает у людей пожилого возраста, чаше у женщин. Не рецидивирует и не метастазирует. Опухоль плотная, серовато-белого цвета. Может быть двусторонней, может инфильтрировать периост лопатки и прилежащих ребер. Многими авторами описывается как мягкий, уплощенный узел, часто трудно отличимый от липомы.
Микроскопически опухоль состоит из бедной клетками фиброзной ткаин с большим количеством волокон и с примесью тонких прослоек жировой ткани. Волокна в опухоли толстые, оксифильиые, частью ШИК-положительные, дают резко выраженную реакцию при окраске на эластику. Каждое такое волокно окружено нежными аргирофильными волоконцами. Волокна имеют структуру пружины, флюоресцируют в ультрафиолетовом свете, перевариваются эластазой.
По А.Р. Stout, в динамике эластофибромы можно отметить ряд стадий первоначально происходит активация фибробластов в ответ на повреждение, затем образуются тонкие ветвистые волоконца, в дальнейшем они импрегируются материалом, дающим реакцию иа эластин, позже появляются оболочки из коллагена, а сами волокна под действием мышечной тяги выпрямляются Часть авторов расценивают эластофиброму как дегенеративный процесс в сочетании с регенеративным тумороподобным процессом. При ультраструктурном исследовании в ряде случаев эластический материал был обнаружен интраиеллюлярно, что позволило высказать соображение о нарушении синтеза промежуточной субстанции фибробластоподобными клетками.

Некоторые авторы расценивают эластофиброму как реактивный процесс, возможно связанный с травмой. Она наблюдается, главным образом, у лиц тяжелого физического труда. Вопрос о природе волокон в эластофиброме обсуждается, предполагают, что они состоят из эластина или же речь идет об «эластической дегенерации коллагена.
Фиброматоз. Этот термин часто употребляется неоднозначно В настоящее время наиболее правильно рассматривать фиброматоз как диспластический процесс соединительной ткани, своеобразное нарушение ее развития в процессе эмбриогенеза и в постнатальном периоде. Морфологически это выражается в пролиферации фибробластических элементов, часто с инфильтрацией окружающих тканей. Фиброматоз не имеет ни признаков процесса воспалительного происхождения, ни черт определенной опухоли, встречается во все периоды жизни в любом месте, может быть локализованным и генерализованным. Общими для всех фиброматозов являются тенденция к рецидивам после хирургического лечения, способность спонтанно регрессировать, осложняться контрактурами, а иногда сочетаться с болезнями других органов.
Многогранность и своеобразие клинико-морфологических проявлений фиброматоза позволяют считать его наиболее частым видом так называемых псевдосаркоматозных поражений.
Значительную роль в изучении и расширении представлений о фиброматозах сыграли работы S. Kauffman, A. P. Stout (1965). F. M. Enzinger (1965), D. Н Mackenzie (1970), Р W Allen (1977), М Larregue и соавт. (1980), в которых рассматривались и вопросы классификации. Наиболее правильным в настоящее время признано разделение фиброматозов на 2 вида врожденные и ювенильные; смешанные. Среди врожденных и ювенильных рассматривают врожденный фибросаркомоподобный фиброматоз, врожденный генерализованный фиброматоз, врожденный мультицентричный фиброматоз врожденный локализованный фиброматоз. фиброматоз шеи, диффузный инфантильный фиброматоз. фиброзную гамартому младенцев, рецидивирующую фиброзную опухоль пальцев, ювеннльную ангиофиброму носоглотке, наследственный гингивальный фиброматоз, множественный ювенильный гиалиновый фиброматоз.
Смешанные формы фиброматозов подразделены на 2 подгруппы, фиброматозы типа Дюпюитрена и фиброматозы десмоидного типа. К фиброматозам типа Дюпюитрена отнесены ладонный, подошвенный и фиброматоз полового члена, к фиброматозам десмоидного типа абдоминальный, экстраабдоменальный и интраабдоминальный.
Читайте также:
- Непарная вена. Полунепарная вена. Топография непарной и полунепарной вены.
- Фибрилляция предсердий
- Электрокардиограмма при декстрокардии и декстроверсии. ЭКГ при декстроверсиях сердца
- Фармакологические пробы при коронарной недостаточности. Предынфарктное состояние
- Тонкокишечная непроходимость при болезни Крона.