Болезнь Крейтцфельдта-Якоба на МРТ головного мозга
Добавил пользователь Skiper Обновлено: 27.01.2026
ФГБОУ ВО «Волгоградский государственный медицинский университет» Минздрава России, Волгоград, Россия
Случай семейной формы болезни Крейтцфельдта—Якоба
Семейная форма болезни Крейтцфельдта—Якоба относится к группе редких тяжелых нейродегенеративных заболеваний, связанных с накоплением в организме патологического прионного белка. Причина болезни генетически детерминирована. Заболевание имеет неуклонно прогрессирующее течение, заканчивается летально в 100% случаев. В клинической картине доминируют симптомы деменции. Нами представлен клинический случай подостро развившейся деменции у 32-летнего пациента. Заболевание имело прогредиентное течение. После обследования у неврологов, генетиков, психиатров и динамического наблюдения был установлен предварительный диагноз «семейная форма болезни Крейтцфельдта—Якоба» на основании критериев Центра профилактики и лечения заболеваний (Central for Desease Control and Prevention, CDC): наличие прогрессирующей деменции, экстрапирамидные расстройства, отсутствие характерных комплексов на электроэнцефалограмме, длительность заболевания менее 2—3 лет, подтвержденная генетическая мутация в гене белка PrP, билатеральные гиперинтенсивные сигналы в области хвостатых ядер, таламуса на Т2-взвешенных изображениях (симптом «медовых сот»), атрофия коры больших полушарий и мозжечка на диффузионно-взвешенной магнитно-резонансной томографии головного мозга (режим DWI). Пациент скончался через 3 года после начала заболевания. В биоптатах были обнаружены патоморфологические признаки спонгиоформной дегенерации нейронов, что является характерным для данного заболевания.
Болезнь Крейтцфельдта—Якоба (БКЯ) относится к редким нейродегенеративным заболеваниям из класса прионных болезней человека [1—3]. Ежегодно регистрируется с частотой 1—2 случая на 1 000 000 в общемировом масштабе. Помимо аномального прионного белка, накапливающегося в мозговой ткани, БКЯ характеризуется спонгиоформными изменениями нервной ткани, гибелью нейронов и глиозом [4]. Неизбежно наступает летальный исход. Трудности прижизненной диагностики БКЯ, вероятно, связаны с отсутствием настороженности и низкой информированностью врачей об этой редкой патологии [5].
Наиболее часто встречающаяся форма БКЯ — спорадическая (85—90%); кроме того, существуют семейная, ятрогенная и вариантная формы [6].
Семейная форма заболевания связана с мутацией в гене, находящемся на коротком плече 20-й хромосомы (локус PRNP, кодоны 178, 200, 210), кодирующим прионный белок PrP аппарата Гольджи с неясной функцией. В результате генетической «поломки» происходят его трансляция и конформация в патологический белок PrPSc с приобретением новых свойств [7].
PrPSc устойчив к действию протеаз и обладает способностью к спонтанной агрегации с образованием палочкообразных или фибриллярных частиц [8]. Таким образом формируется спонгиоформная дегенерация нейронов с появлением внутриклеточных вакуолей, кистообразных расширений отростков нейронов и их гибелью в области коры, подкорковых образований, ствола с последующим развитием грубого астроцитарного глиоза [3].
Средний возраст начала семейной формы БКЯ (семБКЯ) — 29—33 года. Как правило, продолжительность заболевания составляет 2—3 года.
Основным клиническим симптомом семБКЯ является быстро прогрессирующая деменция. Кроме того, могут быть признаки очагового поражения нервной системы: миоклонус, нарушения зрения, мозжечковые (чаще других), пирамидные и/или экстрапирамидные расстройства [3]. Когнитивные нарушения постепенно прогрессируют до появления акинетического мутизма в терминальной стадии [5].
На диффузионно-взвешенной (DWI) магнитно-резонансной томографии (МРТ) головного мозга выявляются аномальные билатеральные гиперинтенсивные сигналы на Т2-взвешенных изображениях (симптом «медовых сот») преимущественно в области головок хвостатых ядер, таламуса, отмечается атрофия коры больших полушарий и мозжечка [6].
Клинический случай. Пациент Ф., 35 лет, поступил в Волгоградскую областную психиатрическую больницу № 2 в конце 2013 г. в сопровождении жены и отца. Активных жалоб не предъявлял. Со слов родственников, у больного отмечались нарушение памяти, частичная дезориентация в пространстве, времени, в собственной личности, потеря интереса к любым происходящим событиям в жизни, отсутствие эмоций.
Данные симптомы появились подостро в 2013 г. и постепенно нарастали. Пациент перестал справляться с домашними обязанностями, повседневными навыками, забывал выключать воду, свет, газ, оставлял покупки в магазине, не помнил дорогу домой. Родственники отрицали наличие у него миоклонических подергиваний, тремора, визуальных, слуховых, сенсорных галлюцинаций, мозжечковых расстройств. Мужчина не подвергался хирургическим вмешательствам, не имел склонности к аллергическим реакциям, не совершал поездки за пределы Волгоградской области в последние 5 лет, не употреблял сырое мясо и мозги скота.
Наследственность отягощена: в пяти предыдущих поколениях родственники по линии матери страдали заболеванием, которое возникало в возрасте от 28 до 36 лет и у мужчин, и у женщин и проявлялось подостро развивающейся деменцией. Все заболевшие члены семьи умирали через 2—3 года после начала болезни.
Неврологическое обследование выявило наличие амнестической афазии, алексии, агнозии, апраксии, аграфии. Патологических изменений черепной иннервации, двигательной и чувствительной сфер не выявлено. Координация движений и статика не страдали. Походка замедлена. Больной не мог выполнить задания по краткой шкале оценки психического статуса (MMSE). Например, на вопросы «назовите текущий год, месяц, день» он отвечал «не знаю». В связи с тем что на последующие вопросы пациент отвечал так же, тестирование по шкале MMSE было прекращено. Простые команды больной не выполнял.
В психическом статусе: контакту малодоступен, внешний вид неряшливый, говорит, что находится «здесь». Мышление малопродуктивное. Внезапно становился возбужденным, озлобленным, вскакивал, куда-то стремился. Критика к своему состоянию отсутствовала.
Исследование крови пациента патологии не выявило (общий анализ крови, биохимический анализ крови, гормональный состав, анализ на ВИЧ, гепатиты, сифилис, глюкоза крови, концентрация церулоплазмина).
Исследование цереброспинальной жидкости показало нормальное содержание белка, клеток, глюкозы. Не были выявлены антитела к токсоплазме гонди, бартонелле, венерическим возбудителям.
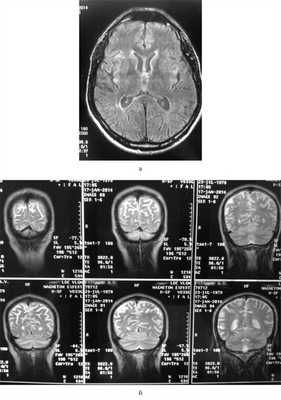
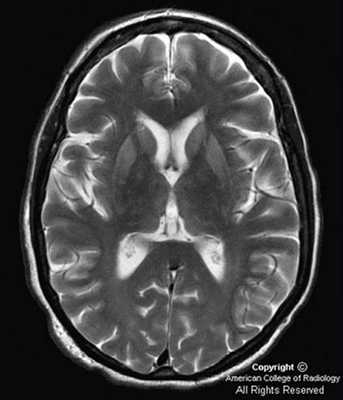
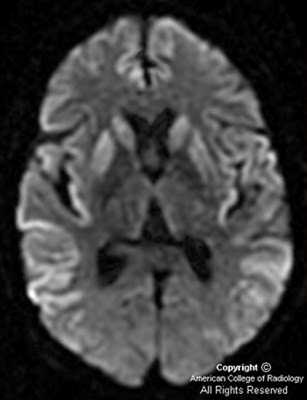
На МРТ головного мозга в режиме DWI были определены аномальные билатеральные гиперинтенсивные сигналы на Т2-взвешенных изображениях, преимущественно в области головок хвостатых ядер, таламуса, атрофия коры больших полушарий (рис. 1). Рис. 1. Билатеральные гиперинтенсивные сигналы в области подкорковых ганглиев на Т2-взвешенных изображениях МРТ головного мозга (а) и атрофия коры головного мозга на МРТ головного мозга на первом году заболевания.
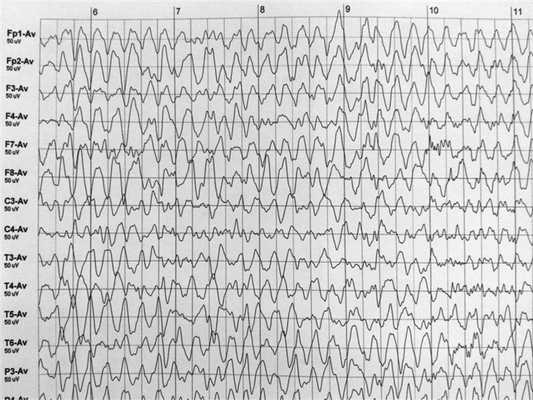
На ЭЭГ было выявлено замедление биоэлектрической активности мозга на фоне дезорганизованного ритма (рис. 2). Рис. 2. Электроэнцефалограмма дезорганизованного ритма с преобладанием медленноволновой активности.
После консультации врача-генетика пациент был направлен на обследование в ФГБНУ «Медико-генетический научный центр», где была выявлена гомозиготность по метионину в 129-м кодоне гена, кодирующего белок PrP, 20-й хромосомы.
С учетом отягощенной наследственности, особенностей появления и развития интеллектуально-мнестических расстройств, отсутствия очаговой неврологической симптоматики, наличия документально подтвержденной мутации в 129-м кодоне гена прионного белка 20-й хромосомы был установлен диагноз: болезнь Крейтцфельдта—Якоба, семейная форма.
С родственниками пациента была проведена беседа о наследственно-обусловленном характере заболевания, которое проявляется в каждом поколении и не зависит от пола. Актуальность информации заключалась в том, что пациент Ф. имел трех несовершеннолетних детей, которые также могут являться носителями данной мутации.
С осени 2013 г. больной был признан инвалидом I группы. Ежегодно госпитализировался в психиатрическую больницу для симптоматической терапии — коррекции когнитивных и поведенческих расстройств.
Во время последней госпитализации в августе 2016 г. на фоне полного соматического благополучия у пациента внезапно появились бледность кожных покровов, непроизвольное мочеиспускание, потеря сознания и затем остановка сердечной деятельности и дыхания. Выполненные реанимационные мероприятия оказались безрезультатными, и была констатирована биологическая смерть.
При последующем патологоанатомическом исследовании были выявлены изменения по типу Штерна—Гарсена (согласно патоморфологической классификации): в стволе головного мозга — очаг кровоизлияния с развитием отека-набухания головного мозга. При гистологическом исследовании обнаружены вакуоли в сером веществе коры головного мозга и подкорковые очаги глиоза, что является патоморфологическим субстратом наследственной прионной болезни — Крейтцфельдта—Якоба — прогрессирующего дистрофического поражения базальных ганглиев головного мозга. При исследовании легких отмечались эмболия мелких ветвей легочной артерии, геморрагические инфаркты в краевых зонах нижних долей обоих легких, признаки острого венозного полнокровия (отек легких, острые язвы слизистой оболочки тела желудка), паренхиматозной дистрофии внутренних органов.
Постановка окончательного прижизненного клинического диагноза стала возможной после генетического анализа в целях выявления мутации в 129-м кодоне гена прионного белка 20-й хромосомы и выявленных изменений на МРТ DWI-томограммах головного мозга в виде аномального свечения хвостатого ядра и скорлупы.
Основные неврологические симптомы семБКЯ, согласно критериям CDC, включают наличие прогрессирующей деменции, экстрапирамидные расстройства, отсутствие характерных комплексов на ЭЭГ, длительность заболевания менее 2—3 лет. Эти симптомы неспецифичны и могут встречаться при других заболеваниях. В связи с этим обновление и расширение базы знаний и диагностических критериев необходимы для проведения дифференциальной диагностики.
На основании критериев семБКЯ данный случаи был оценен как «достоверный», пока пациент был жив. На вскрытии были обнаружены признаки спонгиоформной энцефалопатии, что подтвердило диагноз.
Таким образом, ранняя постановка диагноза семБКЯ представляется крайне важной, так как часто схожие симптомы маскируют вирусные или бактериальные энцефалиты, которые поддаются этиотропному лечению. Несмотря на то что семБКЯ является некурабельным заболеванием, своевременная диагностика позволит пациенту и его семье подготовиться к ожидаемому течению болезни, рассмотреть аспекты ухода.
Болезнь Крейтцфельдта-Якоба ( Коровье бешенство , Синдром кортико-стриоспинальной дегенерации , Спастический псевдосклероз )
Болезнь Крейтцфельдта-Якоба — редко встречающееся дегенеративное заболевание головного мозга, связанное с накоплением в нейронах патологического белка приона. Клинически болезнь Крейтцфельдта-Якоба проявляется слабоумием, пирамидными и экстрапирамидными нарушениями, миоклониями, симптомами поражения мозжечка и нарушением зрения. Диагноз болезни Крейтцфельдта-Якоба основывается на совокупности клинических симптомов, данных ЭЭГ, анализа цереброспинальной жидкости, МРТ и ПЭТ головного мозга, а также морфологического исследования образца тканей мозга, полученного в результате биопсии или посмертно. Эффективное лечение болезни Крейтцфельдта-Якоба пока не найдено. Заболевание имеет 100% летальный исход.
МКБ-10
Общие сведения
Болезнь Крейтцфельдта-Якоба очень редкое заболевание. Ранее в литературе указывалось, что частота его встречаемости примерно 1 случай на 1 млн. человек населения. Однако в 90-х годах прошлого века начали отмечаться случаи так называемого нового варианта болезни Крейтцфельдта-Якоба, связанного с заражением от крупного рогатого скота и названного «коровьим бешенством». Только в Англии за 5 лет от этого заболевания умерло 86 человек. Наиболее распространена болезнь Крейтцфельдта-Якоба среди людей в возрасте 65-70 лет и старше. Однако в новом варианте болезнь Крейтцфельдта-Якоба зачастую поражает лиц молодого возраста. Относительно высокий уровень заболеваемости отмечается в Англии, Израиле, Чили и Словакии.
Причины
Установлено, что болезнь Крейтцфельдта-Якоба имеет инфекционный характер. Заражение может произойти при пересадке зараженных прионами тканей, через нейрохирургический инструмент и препараты крови, при введении некоторых гормональных препаратов (человеческого гонадотропина для лечения бесплодия и соматотропина для терапии гипопитуитаризма). Болезнь Крейтцфельдта-Якоба новой формы может развиваться после употребления в пищу мяса заболевших животных (коровы) или носителей инфекции (овец и коз).
В результате ряда исследований стало известно, что болезнь Крейтцфельдта-Якоба связана с проникновением в организм инфекционного белка — приона. В норме в клетках головного мозга человека содержится здоровый прион, имеющий несколько другое строение. Инфекционный прион, попадая в организм человека не разрушается, а с током крови поступает в головной мозг и откладывается на поверхности нейронов. Его взаимодействие с нормальными прионами мозговой клетки приводит к тому, что они изменяют свою структуру, постепенно трансформируясь в патогенную, подобную инфекционному приону, форму. Патогенные прионы образуют бляшки и приводят к гибели нейрона.
Болезнь Крейтцфельдта-Якоба имеет достаточно длительный инкубационный период, связанный с временем, необходимым для проникновения инфекционных прионов в мозговую ткань и патогенной трансформации здоровых прионов. Длительность инкубационного периода напрямую зависит от способа заражения. При инфицировании тканей головного мозга зараженным хирургическим инструментом болезнь Крейтцфельдта-Якоба развивается через 15-20 месяцев. При инфицировании через имплантированные в околомозговые структуры ткани (например, твердую мозговую оболочку, роговицу глаза) инкубационный период может длиться до 5,5 лет. При внутримышечном введении инфицированных лекарственных препаратов (например, гонадотропина, соматотропина, содержащих бычий тромбин гемостатиков) болезнь Крейтцфельдта-Якоба начинает проявляться спустя 12,5 лет.
Отмечаются также наследственные формы болезни Крейтцфельдта-Якоба, связанные с генетическими нарушениями, приводящими к образованию патологических прионов.
Классификация
Практическая неврология классифицирует болезнь Крейтцфельдта-Якоба с учетом ее клинической формы. В соответствии с этим выделяют: подострую спонгиоформную энцефалопатию, отличающуюся быстрым течением и диффузным поражением мозговой коры; классическую (дискинетическую) форму, представляющую собой сочетание пирамидных и экстрапирамидных симптомов со слабоумием (деменцией); промежуточную форму болезни Крейтцфельдта — Якоба, характеризующуюся преобладанием мозжечковых и подкорковых расстройств; амиотрофическую форму, двигательные и речевые расстройства при которой напоминают клинику бокового амиотрофического склероза.
Различают также наблюдавшуюся ранее спорадическую форму заболевания и новый вариант болезни Крейтцфельдта-Якоба («коровье бешенство»).
Симптомы болезни Крейтцфельдта-Якоба
В большинстве случаев болезнь Крейтцфельдта-Якоба характеризуется постепенным развитием, однако возможно подострое или острое начало. Примерно в 30% случаев болезнь Крейтцфельдта-Якоба начинается с продромальных симптомов: раздражительности, рассеянности, головных болей, нарушений сна, головокружения, ухудшения памяти, снижения зрения, безынициативности, снижения либидо, изменения поведенческих реакций. Возможно эпизодическое возникновение эйфории и/или беспричинного страха, отрывистые бредовые или галлюцинаторные переживания. Из неврологических нарушений в продромальном периоде наблюдаются: шаткость во время ходьбы, парестезии, расстройство высших функций коры головного мозга (алексия, акалькулия и пр.). Описаны несколько случаев, когда болезнь Крейтцфельдта-Якоба дебютировала с появления корковой слепоты.
В стадии развернутых клинических проявлений болезнь Крейтцфельдта-Якоба характеризуется прогрессирующим спастическим параличем (параплегией или гемиплегией), атаксией, эпилептическими припадками. Возникают экстрапирамидные нарушения: мышечная ригидность, атетоз, тремор. Практически у всех больных наблюдаются миоклонии — быстрые неритмичные сокращения отдельных мышц. Чаще всего отмечается миоклонус губы и века. Наблюдаются вторично генерализованные миоклонические приступы. Появляется и нарастает ярко выраженная деменция, сопровождающаяся нарушениями речи вплоть до ее полного распада. Новый вариант болезни Крейтцфельдта-Якоба отличается преобладанием психиатрической симптоматики и расстройств чувствительности. В 100% случаев он сопровождается мозжечковыми нарушениями, в то время как при спорадической болезни Крейтцфельдта-Якоба расстройства функции мозжечка наблюдаются лишь в 40% случаев.
В терминальной стадии болезнь Крейтцфельдта-Якоба характеризуется глубокой деменцией. Пациенты не контактны, находятся в состоянии прострации, утрачен контроль над функцией тазовых органов. Наблюдаются гиперкинезы, выраженные мышечные атрофии, нарушения глотания, пролежни. Возможна гипертермия и эпилептические приступы. Смерть наступает в коматозном состоянии на фоне децеребрационной ригидности и выраженной кахексии.
Диагностика
Клиническая диагностика заболевания основана на сочетании прогрессирующей в течение 2-х лет деменции, пирамидных и экстрапирамидных расстройств, миоклоний, мозжечковых расстройств и нарушений зрения. Для уточнения диагноза невролог назначает инструментальные методы обследования: электроэнцефалографию (ЭЭГ), ПЭТ и МРТ головного мозга, люмбальную пункцию. В сомнительных случаях для установления диагноза болезнь Крейтцфельдта-Якоба производят стереотаксическую биопсию головного мозга.
На ЭЭГ на фоне сниженной биоэлектрической активности у большинства больных наблюдаются периодические или псевдопериодические острые волны. Отмечается билатеральная, фокальная или генерализованная миоклоническая активность, которая в начальной стадии определяется у половины больных, а в терминальной стадии выявляется в 100% случаев спорадической болезни Крейтцфельдта-Якоба. Новый вариант заболевания часто протекает без существенных изменений ЭЭГ-паттерна.
При проведении МРТ головного мозга в Т2-режиме определяется так называемый «симптом медовых сот» - участки повышенного сигнала, исходящие от подкорковых ганглиев и таламуса. Зачастую выявляются признаки атрофических изменений мозжечка и коры головного мозга, расширение желудочков и боковых цистерн мозга. ПЭТ диагностирует зоны пониженного метаболизма, локализующиеся в подкорковых ядрах, полушариях мозжечка и коре мозга.
Люмбальная пункция в обязательном порядке проводится пациентам с подозрением на болезнь Крейтцфельдта-Якоба. Она позволяет оценить давление ликвора и произвести исследование цереброспинальной жидкости. Отсутствие патологических изменений ликвора позволяет дифференцировать болезнь Крейтцфельдта-Якоба от многих других заболеваний ЦНС.
Наиболее достоверным методом диагностики является морфологическое исследование образцов мозговой ткани, которые могут быть получены прижизненно путем биопсии или при аутопсии после смерти пациента. Применение иммуноцитохимического метода позволяет обнаружить в исследуемом материале отложения патологического белка — приона.
Дифференциальная диагностика
Дифференциальную диагностику болезни Крейтцфельдта-Якоба необходимо проводить с лобно-височной деменцией, герпевирусным энцефалитом, болезнью Альцгеймера, мультиинфарктной деменцией (слабоумием, развивающимся после повторных ишемических и геморрагических инсультов), хроническим менингитом, арахноидитом, нормотензивной гидроцефалией, энцефалопатией Хашимото, сопровождающей некоторые случаи аутоиммунного тиреоидита, и др.
Лечение болезни Крейтцфельдта-Якоба
В современной медицине подходы к лечению болезни Крейтцфельдта-Якоба находятся в стадии активной разработки. Общепринятые противовирусные препараты, а также пассивная иммунизация и вакцинация людей и животных выявились неэффективными. Отмечено, что блокирующее действие на синтез патологических прионов в инфицированных нейронах оказывает Брефелдин А, а блокаторы кальциевых каналов продлевают жизнь инфицированных клеток. Обычно пациенты, имеющие болезнь Крейтцфельдта-Якоба, получают симптоматическое лечение. Оно направлено на купирование миоклонических приступов и экстрапирамидных нарушений, в связи с чем применяются антиэпилептические и противопаркинсонические лекарственные средства.
Прогноз болезни Крейтцфельдта-Якоба
Болезнь Крейтцфельдта-Якоба является фатальным заболеванием. Продолжительность жизни большинства больных не превышает 1 год с момента начала клинических проявлений; а средняя длительность составляет 8 месяцев. Лишь 5-10% заболевших живут в течение 2 и более лет. Наследственная болезнь Крейтцфельдта-Якоба в среднем длится около 26 месяцев.
Болезнь Крейтцфельдта-Якоба на МРТ головного мозга
Заболевания
Болезнь Крейтцфельдта-Якоба
Это быстро прогрессирующее фатальное нейродегенеративное заболевание, ключевым патогенетическим звеном которого является гибель нейронов, индуцированная прионовыми белками. Заболевание приводит к быстро прогрессирующему слабоумию и смерти обычно в течение года от начала.
История
Заболевание было названо в честь Ханса Герхарда Кройцфельдта (1885-1964), немецкого невролога, который впервые описал это состояние в 1920 году, и Альфонса Марии Якоба (1884-1931), немецкого невролога, которая также описала это состояние в отдельном исследовании в 1921 году.
Эпидемиология
Описаны три основных типа болезни Крейтцфельдта-Якоба:
- Спорадический - составляет 85-90% случаев и разделена на множество подтипов в зависимости от мутации;
- Семейный - 10% случаев, вызвана мутацией PRPc;
- Ятрогенный;
Особенности клиники
К основным клиническим проявлениям заболевания относятся: быстро прогрессирующие когнитивные нарушения, миоклонус, дистония, акинетикоригидный синдром, спастичность, гиперрефлексия, атаксия, зрительные расстройства, на поздних этапах заболевания акинетический мутизм. Примерно в трети случаев отмечаются эпилептические припадки. Средняя продолжительность жизни при спорадической форме БКЯ составляет около 5 мес; более 90% пациентов умирают в течение 1 года из-за аспирационной пневмонии в состоянии акинетического мутизма. Используемые в настоящее время диагностические критерии БКЯ включают быстро прогрессирующую деменцию, экстрапирамидно/ пирамидные и зрительные расстройства, миоклонус, мозжечковую атаксию, а также характерные изменения ЭЭГ в виде комплексов высокоамплитудных 2-3-фазных острых волн и положительный маркер белок 14-3-3 в цереброспинальной жидкости (ЦСЖ). Сложности диагностики БКЯ связывают с редкостью заболевания, клиническим полиморфизмом и недостаточной информированностью врачей.
До недавнего времени «золотым стандартом» верификации диагноза БКЯ являлась биопсия головного мозга, позволяющая выявить характерные изменения в мозговой ткани в виде мелких вакуолей в телах нейронов, из-за чего ткань мозга приобретает губчатый вид, пролиферации клеток глии при отсутствии признаков воспаления. При электронной микроскопии возможно обнаружение прионных палочек, являющихся патогномоничным признаком заболевания. Указанные морфологические изменения отмечаются в коре головного мозга, базальных ганглиях, мозжечке и верхних отделах ствола мозга. Однако в случаях БКЯ биопсия мозга не нашла широкого применения в клинической практике из-за инвазивности метода, сложности санитарной обработки оборудования и утилизации биоматериалов, связанных с высокой устойчивостью прионов, а также вследствие небольшого объема биоптата мозговой ткани, что может быть причиной ложноотрицательных результатов морфологического и иммуногистохимического исследования. Однако в настоящее все больше распространена МРТ диагностика данного заболевания, по причине выявления специфических изменений, наиболее информативными являются последовательность DWI.
МРТ при болезни Крейтцфельдта-Якоба
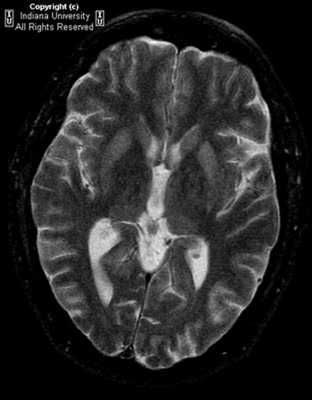
Болезнь Крейтцфельдта-Якоба - редкое дегенеративное заболевание центральной нервной системы, при котором происходит поражение коры и базальных ганглиев головного мозга, а также нарушение функций спинного мозга.
Болезнь Крейтцфельдта-Якоба (она же - губчатая энцефалопатия) встречается довольно редко и может длительное время протекать бессимптомно. Длительность инкубационного периода зависит от способа заражения: так, с момента заболевания до появления первых симптомов может пройти от 12 месяцев до 10-15 лет. Источником болезни, по распространенному мнению, считается коровье мясо, зараженное прионами*. Однако болезнь может носить наследственный характер или может быть занесена во время операции нестерильными медицинскими инструментами.
Особенности развития болезни Крейтцфельдта-Якоба
Как правило, болезнь включает три стадии развитии: продромальный период, инициальный и развернутый. Первая стадия (продромальная) характеризуется быстрой утомляемостью пациента, расстройством сна, отсутствием аппетита и прочими признаками, которые могут быть приняты, например, за проявление депрессии. Через несколько недель или месяцев наступает второй этап (инициальный), когда пациент испытывает головокружения и головные боли, ощущает ухудшение зрения.
Третья стадия, развернутая, может привести к спастическому параличу и таким формам сокращения мышц, как миоклония, тремор, ригидность. На этом этапе у пациента происходит атрофия нейронов головного мозга. Кроме того, пациент постоянно чувствует напряженность в мышцах. При некоторых клинических формах болезнь на третьей стадии сопровождается дисфункцией мозжечка.
Для достоверной диагностики данного заболевания наиболее эффективным методом является прижизненная биопсия с забором вещества мозга. Однако, как правило, биопсия назначается как крайний метод диагностики. МРТ в данном случае позволяет осуществить дифференциальную диагностику, что поможет специалисту исключить большинство различных патологий.
Болезнь Крейтцфельдта-Якоба: МРТ-диагностика
Для диагностирования дегенеративного заболевания лечащий врач может назначить комплекс взаимоуточняющих методик: ЭЭГ и МРТ головного мозга.
Магнитно-резонансная томография позволяет получить качественные и контрастные изображения головного мозга пациента. Во время проведения МРТ выявляются участки повышенного сигнала, локализованные в подкорковых ганглиях и таламусах. Кроме того, магнитно-резонансная томография дает возможность успешно определять признаки атрофических изменений мозжечка, коры головного мозга и расширение желудочковой системы.
МРТ является абсолютно безвредным и безболезненным методом диагностики. МРТ является абсолютно безвредным и безболезненным методом диагностики. Высокое контрастное разрешение позволяет распознавать любую патологию структур головного мозга, оценить состояние белого и серого вещества, черепно-мозговых нервов, ствола мозга, образований задней черепной ямки.
Магнитно-резонансная диагностика часто назначается в целях дифференциальной диагностики заболеваний нервной системы. Как мы уже отметили, болезнь Крейтцфельдта-Якоба встречается довольно редко и трудно идентифицируется, поэтому при постановке такого диагноза необходимо исключить другие заболевания головного мозга. Ими могут быть лобно-височная и мультинфарктная деменция, энцефалит, хронический менингит, нормотензивная гидроцефалия, энцефалопатия и другие заболевания, некоторые из которых, имеющие схожую с болезнью Крейтцфельдта-Якоба клиническую картину, вызывают деменцию. У пациентов с расстройством интеллекта (слабоумием) МРТ также выявляет структурные изменения в головном мозге и его аномалии.
Таким образом, при диагностике болезни Крейтцфельдта-Якоба важным преимуществом МРТ (в сравнении с другими методами лучевой диагностики) является возможность исключить большинство различных патологий.
Позвонив сейчас, даже если у вас не стоит остро вопрос об оказании психиатрической помощи или лечения — вы однозначно получите развернутую консультацию, содержащую основные правила оказания этой помощи, информацию об эффективности современных методик, а также ответы на все вопросы. Обладая всей информацией по столь щекотливой и важной проблеме, мы гарантируем, что вы не ошибетесь, когда придет время действовать быстро.
Тем более, необходимо звонить, если нужна
экстренная помощь
Проверял Шайдуллин Ренат Флюрович
Болезнь Крейтцфельдта-Якоба относится к редким видам дегенеративных поражений головного мозга. Она входит в группу прионных трансмиссивных энцефалопатий, которые вызываются попаданием в организм аномальной изоформы белка приона и его накапливание в нейронах.

Патология возникает у одного из миллиона человек на земном шаре. Заболевание было впервые описано в 1920 году германским невропатологом Гансом Кройцфельдтом. А спустя год его земляк и коллега Альфонс Якобс уточнил, что для отклонения характерно проявление психических нарушений на фоне органического поражения пирамидальной, экстрапирамидальной области ЦНС, передней части рогов спинного мозга, мозжечка и нарушениями зрения, связанное с изменениями корковых структур.
Частота возникновения этой патологии невелика - она регистрируется у одного человека на миллион. В 90 годах прошлого столетия стали отмечаться симптомы нового варианта болезни Крейтцфельдта-Якоба, которые имели инфекционную природу и связывались с заражением от коров и другого крупного рогатого скота («коровье бешенство»). Чаще всего заболевание регистрируется у людей в возрасте от 65 лет, но в последние годы отклонения такого типа отмечаются и у более молодых пациентов.
Что вызывает синдром Крейтцфельдта-Якоба
Заболевание вызывается наличием в организме особого белка (приона), который имеет нарушенную структуру и оказывает негативное влияние на нервную систему. Действие приона напоминает эффект вирусов, вызывающих гибель клетки при своем размножении, но при этом он не несет в себе генетический материал (ДНК или РНК).
В своем стремлении избавиться от агрессора, клетка, которая подвергается атаке, начинает активно выделять вещества. Но белок, осевший на мембране, не позволяет им выйти наружу, и они разрушают органеллы и запускают процесс апоптоза. Соседние структуры начинают реагировать на это воспалением, в связи с чем и происходит дальнейшее поражение клеток.
Прионы выдерживают большой температурный диапазон, облучение, они устойчивы к ферментативному расщеплению. На них не оказывают влияния антибиотики и противовирусные средства. Под воздействием этого белка ткань нервной системы становится «дырявой» и начинает напоминать губку.
Заражение экзогенным путем (помимо спонтанной внутренней трансформации прионов и наследственной формы заболевания) происходит из внешней среды такими способами:
- При употреблении пищи животного происхождения. Особенно много инфекционного агента «коровьего бешенства» наблюдается в говядине, мозге, крови и селезенке. Описываются случаи заболевания у народов, которые практикуют ритуальный каннибализм.
- При пересадке внутренних органов, в том числе печени, почек, роговицы, сердечной мышцы. Возможность передачи сохраняется при переливании как цельной крови, так и ее отдельных компонентов (массы эритроцитов, лейкоцитов, плазмы).
- Применение препаратов, полученных их ткани животных.
- Передача от человека к человеку через биологические жидкости путем попадания их на раневые поверхности или в желудок.
Болезнь Крейтцфельдта-Якоба не передается воздушно-капельным путем, как это бывает при вирусных инфекциях, а также обычным прикосновением. Есть предположения передачи патологического белка приона через мокроту, мочу и кал, но исследований этого варианта заражение проводилось недостаточно.
Патогенез болезни
В настоящее время выделяется два типа патогенеза болезни Крейтцфельда - Якоба. Первый - экстрацеребральный, который не относится к мозговым структурам и церебральный. Оба типа возникают при заражении прионами извне, а наследственная форма заболевания и спорадическая до сих пор изучены недостаточно.
Внецеребральный вариант развития патологии заключается в том, что извращенный белок поступает в организм и соединяется с лимфоцитами, макрофагами и другими клетками, участвующими в иммунной защите. Он переносится ими в ткани лимфатической системы (селезенку, лимфоузлы). Там происходит его размножение, причем организм не препятствует этому, так как не воспринимает прион как чужеродное тело. При этом он не повреждает и не вызывает увеличения лимфоидных органов, а только размножаются и сохраняются в них.
В дальнейшем распространение патогена происходит под влиянием вегетативной нервной системы. Они направляются в центральную нервную систему, и начинается церебральный этап болезни Крейтцфельда - Якоба. Они накапливаются во внутриклеточной жидкости и нервных окончаниях. В результате происходит снижение нормального прионного белка, который участвует в обычном питании клеток.

Классификация болезни Крейтцфельдта-Якоба
В зависимости от особенностей симптоматики, при болезни Крейтцфельдта-Якоба различают следующие формы:
- подострая энцефалопатия с диффузным поражением ткани и быстрым течением;
- дискинетическая, или классическая (сочетание экстрапирамидальных и пирамидальных нарушений с проявлениями деменции);
- промежуточная с преобладанием симптомов поражения мозжечка и областей подкорки;
- амиотрофическая (с нарушением речи и движений).
По этиологии болезнь Крейтцфельдта-Якоба подразделяется на следующие виды:
- Спонтанная. Второе название - классическая. Прионы в этом случае не попадают в организм извне, а формируются без видимой причины. Скорее всего, современная медицинская наука просто не может найти объяснением этому явлению. Чаще страдают люди старшего возраста (после 50 лет).
- Наследственная. При таком отклонении формирование приона обусловлено генетически.
- Ятрогенная. Повреждающий белок при данном виде болезни попадает в организм извне. Его источником ранее могли служить некоторые препараты. Большая часть из них в настоящее время не используются. Причиной также выступают оболочки, взятые из ткани трупов для закрытия ран при операциях на мозге.
- Новая форма болезни Крейтцфельдта-Якоба. Впервые описана медиками Великобритании в 1995 году. Причиной ее стало употребление мяса «бешеных коров» с наличием прионов. В отличие от классического варианта патология наблюдается людей в возрасте от 20 лет и проявляется в виде личностных изменений.
Болезнь Крейтцфельдта-Якоба: симптомы
Болезнь Крейтцфельдта-Якоба имеет характерную симптоматику, независимо о происхождения. Ятрогенная, наследственная и классическая форма проявляется примерно одинаково. Несколько иначе протекает новый вид заболевания.
Основные проявления
Начальная стадия проявляется временными выпадениями из памяти событий, изменением настроения, потерей интереса к окружающим. Постепенно пациент испытывает все больше трудностей в осуществлении жизненно необходимых действий. Конечная стадия сопровождается нарушением зрения, медленной речью и появлением галлюцинаций.
Примерно у 40% больных со спорадической формой болезнь протекает в подостро, с постепенно усугубляющимися когнитивными отклонениями. Еще 40% зараженных страдают нарушениями функций мозжечка, а у остальных описывается смешанная форма заболевания.
Наиболее характерными клиническими симптомами классической болезни Крейтцфельдта-Якоба являются:
- нарушение поведения; ;
- диплопия и развитие корковой слепоты; ;
- дисфункция мозжечка, атаксия;
- нарушение координации;
- парезы;
- дизартрия;
- неспособность к самообслуживанию, неряшливость и другие признаки слабоумия;
- сочетание признаков поражения пирамидной и экстрапирамидной системы.
У многих пациентов отмечаются локальные или генерализованные виды эпилептических припадков. Они проявляются в ответ на определенные раздражители - свет, звук, прикосновение.
Терминальная стадия сопровождается глобальными когнитивными отклонениями, полной неспособностью к передвижению. Смерть обычно наступает в течение от 8 месяцев до 2 лет после появления начальных отчетливых симптомов заболевания. Новая форма болезни характеризуется выходящими на первый план психическими нарушениями и изменениями чувствительности.
Стадии
Специалисты выделяют три стадии протекания болезни Крейтцфельдта-Якоба:
- Продромальная стадия. Симптомы ее не имеют специфических проявлений и наблюдаются примерно у 30% всех заболевших. У человека отмечается астения, нарушение сна и аппетита, снижение способности к запоминанию, похудение, легкие когнитивные отклонения. Для пациента характерно появление фобий, бреда, галлююцинаций.
- Инициальная стадия. Начинаются изменения со стороны зрения, появляется головная боль, нарастает неустойчивость, атаксия, нарушение чувствительности, тремор, миоклонии, эпилептические припадки, и другие неврологические симптомы на фоне проявления слабоумия.
- Развернутая стадия сопровождается спастическим параличом, всеми признаками нарушений работы мозговых структур, тяжелой формой деменции без возможности контакта с больным. У человека полностью утрачивается контроль над работой тазовых органов, нарушается глотание, отмечается снижение температуры.
Чаще всего болезнь Крейтцфельдта-Якоба развивается постепенно, но иногда она имеет острое и подострое начало. Смерть наступает в результате отказа от работы жизненно важных центров и выраженного истощения.
Диагностика болезни Крейтцфельдта-Якоба
Предположительный диагноз болезни Крейтцфельдта-Якоба ставится на основании клинических проявлений и данный дополнительных методов исследования. Заподозрить патологию можно при сочетании определенных симптомов, наблюдающихся в течение продолжительного времени:
- прогрессирование деменции;
- пирамидальные и экстрапирамидальные нарушения;
- миоклонии;
- расстройства работы мозжечковых структур;
- нарушение зрения.
Для уточнения патологии невролог направляет человека на прохождение энцефалографии, МРТ мозга, спинномозговую пункцию, стереотаксическую биописию:
- На ЭЭГ у почти у каждого пациента с болезнью Крейтцфельдта-Якоба отмечается снижение электрической активности с периодически выскакивающими острыми волнами. Наблюдаются изменения, характерные для миоклонии, у 50% больных. Новый вид заболевания часто не проявляется какими-либо нарушениями на энцефалограмме.
- На МРТ можно выявить признаки атрофических изменений в мозжечке и коре мозга (синдром «медовых сот»), расширение желудочков. От подкорковых структур и таламуса иногда улавливаются участки с повышенным сигналом.
- Люмбальная пункция проводится в обязательном порядке при подозрении на болезнь Крейтцфельдта-Якоба. Исследование ликвора позволяет провести дифференциальный диагноз с другими патологическими нарушениями ЦНС.
- Самым объективным способом, который помогает достоверно определить диагноз, является морфологическое исследования биоптата ткани мозга. С помощью иммунохроматографического метода есть возможность обнаружить белок прион. Его наличие позволяет утверждать о том, что у больного наблюдается именно болезнь Крейтцфельдта-Якоба.
Проводить дифференциальную диагностику приходится с другими заболеваниями центральной нервной системы, которые протекают с похожими симптомами:
- старческая, мультиинфарктная деменция; ;
- энцефалопатия Хашимото;
- воспалительное поражение мозговых оболочек (энцефалит, арахноидит, менингит).
Лечение болезни Крейтцфельдта-Якоба в Москве
Этиотропная терапия заболевания до сих пор находится в разработке. Поэтому все лечение его в клинике направлено на купирование симптоматических проявлений. Пациенту отменяются все ранее применяемые препараты, поскольку они могут негативно отражаться на течении болезни, усугубляя нарушение мнестических функций и ухудшая поведение больного.
Консервативная терапия
Уже доказано, что традиционные средства, которые успешно используются при лечении вирусной инфекции (вакцины, интерфероны) не оказывают положительного действия на течение болезни Крейтцфельдта-Якоба. Некоторые результаты с временным улучшением состояния удается получить при введении антибиотика, который блокирует работу аппарата Гольджи внутри клетки и препятствует размножению прионов. Замечено, что при использовании некоторых блокаторов каналов кальция длительность функционирования пораженных структур мозга увеличивается по времени.
Некоторое замедление накопления патологических белков отмечаются при использовании иммуномодулятора, который стимулирует выработку эндогенного интерферона. Но эти препараты обладают сильными побочными действиями.
Симптоматическая терапия у больных направлена на устранение:
- тремора и приступов эпилепсии;
- экстрапирамидальных нарушений; и беспокойства;
- болевого синдрома.
Для этого используются различные группы препаратов, их выбор осуществляет врач. Основанием к применению того или иного средства является наличие и степень выраженности отклонения. В терапии применяются такие группы препаратов:
- нейролептики;
- транквилизаторы;
- анальгетики;
- опиаты;
- седативные средства;
- антидепрессанты;
- противосудорожные лекарства.

Преимущество нахождения больного в клинике
После установления диагноза болезни Крейтцфельдта-Якоба, человеку требуется обеспечить полноценную симптоматическую терапию, психологическую поддержку пациента и членов его семьи. Чем дальше прогрессирует патология, тем больше необходимо пациенту заботы и внимания, так как у него постепенно утрачиваются навыки самообслуживания, нарушается работа органов малого таза.
Работающие родственники не могут обеспечить круглосуточное дежурство возле пациента. А его потребуется кормить, мыть и помогает в передвижении по мере необходимости. В некоторых случаях ему приходится выводить мочу при помощи катетеризации в результате нарушения работы мочевого пузыря. А эту манипуляцию в состоянии выполнить только специалисты с медицинским образованием.
При нарушении функции глотания человеку потребуется питания при помощи специальной трубки и постановка капельных растворов с необходимыми веществами. В домашних условиях обеспечить полноценное лечение и уход возможно только на самых ранних стадиях. В последующем больному обязательно потребуется систематический уход специально подготовленного персонала.
В терминальной стадии проводятся мероприятия направленные на продление жизни. Их осуществляют только при наличии реанимационного оборудования - ИВЛ, оксигенотерапия, средства для стимуляции работы дыхательной и сердечно-сосудистой системы.
При выборе клиники для оказания помощи пациентам с болезнью Крейтцфельдта-Якоба, родственникам следует понимать, что наиболее качественная помощь может быть оказана в заведении, в котором работают опытные специалисты с определенным профилем.
Если госпитализировать человека в государственный стационар, то можно сэкономить средства, но при этом условия пребывания будут не идеальными:
- в палатах размещаются по нескольку человек;
- препараты придет приобретать самостоятельно, так как те, что есть в наличии не всегда могут эффективно устранять осложнения;
- у пациента есть высокий риск подхватить внутрибольничную инфекцию;
- персонал не сможет постоянно контролировать пациента и оказывать ему помощь при необходимости 24 часа в сутки.
Когда больной находится еще в сознании и способен оценивать сложившуюся ситуацию, его пребывание в госучреждении значительно усугубит состояние, вызовет депрессию и усиление психических нарушений.

Вопросы и ответы
Если находится в постоянном контакте с человеком, у которого болезнь Крейтцфельдта-Якоба, есть ли шанс заразиться?
Заражение при обычном контакте происходит крайне редко. Но во время ухода следует придерживаться гигиенических правил. Они заключатся в мытье рук перед едой, защите кожи от попадания биологических жидкостей, закрытии всех раневых поверхностей водонепроницаемыми повязками.
Можно ли вылечить человека при этом заболевании?
Данное заболевание не поддается лечению и приводит к летальному исходу со 100% степенью вероятности. Все что можно предпринять в этом случае - обеспечить достойный уход за пациентом, купировать возникающие осложнения и проводить симптоматическую терапию с целью повышение качества жизни.
Почему болезнь Крейтцфельдта-Якоба приводит к смерти?
По мере прогрессирования патологии развиваются различные осложнения, которые чаще всего становятся причиной смерти. Некоторые из них поддаются медикаментозной коррекции - эпилептические приступы, острые психозы, пневмонии из-за присоединения инфекции. Правильный уход может снизить страдания человека от появления пролежней в результате дегенерации мышц. Финалом заболевания становится развитие дыхательной или сердечно-сосудистой недостаточности, которые заканчиваются летальным исходом. Но даже эти функции можно поддерживать еще некоторое время с помощью специальных приборов.
Читайте также: