Клиника болезни Гоше - клинические формы
Добавил пользователь Евгений Кузнецов Обновлено: 22.01.2026
В лаборатории наследственных болезней обмена веществ ФГБНУ «Медико-генетический научный центр имени академика Н.П. Бочкова» проводится программа диагностики и контроля лечения болезни Гоше (БГ).
Критерии включения в программу
- Гражданство Российской Федерации
- Любой возраст
Наличие более одного из клинических симптомов/изменений лабораторных показателей для болезни Гоше
- Задержка физического развития
- Гепатоспленомегалия, преимущественно спленомегалия
- Тромбоцитопения, анемия
- Спленэктомия в анамнезе
- Носовые кровотечения, экхимозы
В программу принимаются образцы пациентов с установленным ранее диагнозом болезни Гоше для проведения контроля терапии.
Материал для анализа
Необходимые документы
Подробнее о программе
В рамках программы проводится исследование активности бета-глюкоцереброзидазы, а также высокоспецифичных метаболитов характерных для болезни Гоше (lysoGbl1) методом тандемной-масс спектрометрии. Генетическая (поиск мутаций в гене GBA) и дополнительная биохимическая диагностика проводится при выявлении отклонений от нормы по результатам первичной биохимической диагностики.
Для вызова курьера, для осуществления транспортировки биологического материала из других городов, звоните по телефону:
Время работы горячей линии с 10:00 до 19:00 по московскому времени.
Горячая линия предназначена исключительно для медицинских работников.
Также биоматериал и документы можно доставить самостоятельно по адресу: г. Москва, ул. Москворечье д.1, кабинет 235 (регистратура ФГБНУ «МГНЦ»)
Дополнительная информация
Сроки проведения биохимической диагностики - 14 рабочих дней, ДНК-диагностики от 25-45 рабочих дней.
В ФГБНУ «МГНЦ» с 10:00 до 19:00 по московскому времени в рабочие дни работает горячая линия для врачей 8 800 201 09 39.
Болезнь Берже ( IgA-нефропатия , Идиопатическая возвратная гематурия , Очаговый пролиферативный гломерулонефрит , Синфарингитная гематурия )
Болезнь Берже - это форма гломерулонефрита инфекционно-аутоиммунного характера, характеризующаяся мезангиопролиферативным воспалением с отложением иммунных комплексов. Клиническое течение сопровождается периодической макрогематурией вскоре после инфекционных заболеваний дыхательных путей, органов ЖКТ, изредка возможна постоянная микрогематурия, протеинурия, нефротический синдром с перемежающейся ОПН. Диагностика основывается на результатах лабораторных исследований мочи, иммунограммы крови, гистологического изучения биоптата почки. В лечении патологии используют симптоматическую терапию, иммуносупрессивные средства, антигипертензивные препараты.
Общие сведения
Болезнь Берже (IgA-нефропатия) также известна под названиями очаговый пролиферативный гломерулонефрит, синфарингитная гематурия или идиопатическая возвратная макрогематурия. Синонимические названия отражают разные черты патологии - пролиферативный характер воспаления почек, рецидивирующее развитие гематурии и ее связь с заболеваниями верхних дыхательных путей (фарингитом). Считается самым распространенным вариантом гломерулонефрита в мире - средняя частота встречаемости составляет 5 случаев на 100 тысяч населения, в азиатских странах регистрируется в 5-6 раз чаще, колеблется от 5 до 30% всех случаев воспаления почек. Мужчины страдают от синдрома Берже в несколько раз чаще женщин, среди заболевших преобладают лица в возрасте от 15 до 30 лет. Наблюдается некоторая сезонность обострений (увеличение в осенне-зимний период), что связано с большей частотой простудных заболеваний в холодное время года.
Причины
Болезнь Берже является патологией со сложной и многофакторной этиологией, включающей в себя ряд инфекционных, иммунологических и генетических особенностей организма. Достоверно доказана взаимосвязь заболевания с рядом бактериальных и вирусных инфекций, некоторыми аутоиммунными патологическими состояниями и определенными генетическими мутациями. Роль генетики в патогенезе состояния косвенно доказывают также особенности этнического и расового распространения болезни. Таким образом, выделяют следующие группы этиологических факторов:
- Инфекционные факторы. К ним относят разнообразные бактериальные и вирусные инфекции дыхательных путей, ЖКТ, кожи. У многих больных в анамнезе часто обнаруживаются ангины, гастрит, вирусный гепатит, герпесвирусная инфекция. Считается, что поражение почечной ткани при этих инфекциях обусловлено нетипичной иммунологической реакцией на антигены возбудителя.
- Иммунологические факторы. Патогенетическим субстратом состояния считается появление иммунных комплексов, которые не могут элиминироваться печенью и почками. Причиной их образования являются нарушения синтеза разных форм иммуноглобулина А, нетипичная реакция иммунитета на экзогенные (инфекционные, реже аллергенные) и эндогенные (опухолевые, клеточные) антигены.
- Генетические факторы. Этнические особенности распределения синдрома Берже обусловлены генетическими факторами. В частности, у больных обнаружены нетипичные мутации на 6-й хромосоме, изменения в генах, контролирующих синтез цепей главного комплекса гистосовместимости и рецепторов гепатоцитов. Выявлена повышенная частота развития патологии у лиц, имеющих некоторые наследственные заболевания - целиакию, галактоземию и ряд других.
Роль влияния внешних факторов является предметом обсуждения - возможно, они выступают причиной полиморфной клинической картины состояния. Употребление некоторых продуктов питания, характерных для азиатского региона (неочищенный рис, морепродукты), может усугублять течение нефропатии, что наряду с генетическими особенностями становится причиной более частой регистрации заболевания на этой территории. Роль сопутствующих патологий (артериальной гипертензии, воспалений мочевыводящих путей) в развитии болезни на сегодняшний момент не установлена.
Патогенез
В настоящее время известно несколько основных патогенетических сценариев развития IgA-нефропатии. При заболевании в крови возникают иммунные комплексы, содержащие полимерные формы иммуноглобулина А, которые обычно выделяются на слизистых оболочках и в ничтожном количестве встречаются в кровотоке. Выделение повышенных количеств полимерных разновидностей IgA обусловлено аномалиями их синтеза в костном мозге и нарушениями элиминации через печень по причине дефектов в рецепторах гепатоцитов. В конечном итоге единственным путем выведения иммуноглобулинов и связанных с ними антигенов становятся почки, но размер молекул слишком велик, поэтому они могут откладываться в клубочках нефронов.
Отложения иммунных комплексов в тканях почек активируют лейкоцитарную реакцию и систему комплемента, из-за чего возникает вялое диффузное воспаление с попаданием в мочу эритроцитов и незначительных количеств белка. Влияние факторов, активирующих синтез иммуноглобулинов (воспалительные реакции, инфекционные заболевания), приводит к попаданию больших количеств IgA в выделительную систему и усиливает иммунные реакции. Именно с этим связаны обострения IgA-нефропатии вскоре после перенесенных ангин, ларингитов, заболеваний ЖКТ, вирусных инфекций. Выраженность воспалительных проявлений в период обострения может быть высокой, способна приводить к преходящей острой острой почечной недостаточности. Скрытее и вялотекущие формы могут протекать бессимптомно длительное время, развитие ХПН в большинстве случаев занимает десятки лет.
Классификация
Болезнь Берже подразделяют на несколько клинических форм, которые характеризуются разной тяжестью проявлений, прогностическими данными и подходами к лечению. Разделение является условным - разновидности состояния могут перетекать одно в другое. Это дает повод некоторым исследователям рассматривать их как стадии развития единого патологического процесса, прогрессирующего под действием внешних и внутренних факторов. Значительные различия в скорости прогрессирования приводят к видимости существования различных форм нефропатии. В урологии и нефрологии выделяют три основные формы заболевания:
- Синфарингитная форма. Считается самой распространенной, проявляется периодическими обострениями, которые связаны с воспалительными заболеваниями дыхательных путей или желудочно-кишечного тракта (кишечная инфекция). Пик проявлений возникает через 1-2 дня после инфекционной патологии, их выраженность колеблется от макрогематурии и болей в пояснице до преходящей ОПН. В период между обострениями никаких клинических или лабораторных симптомов нефропатии не определяется.
- Латентная форма. Регистрируется примерно у трети больных, характеризуется менее выраженными симптомами, но считается более серьезной в прогностическом отношении. Обычно никаких субъективных проявлений не обнаруживается, признаки синдрома Берже выявляются при лабораторном исследовании мочи в виде слабой протеинурии и микрогематурии. Количество выделяемого с мочой белка постепенно увеличивается, фильтрующая способность почек уменьшается, что создает условия для развития ХПН.
- Нефротическая форма. Диагностируется у небольшого процента больных с IgA-нефропатией, сопровождается выраженной протеинурией, макрогематурией, онкотическими отеками, гиповолемией и гиперлипидемией. Крайне редко возникает первично, обычно становится осложнением синфарингитных и латентных форм заболевания.
Симптомы болезни Берже
Клинические проявления зависят от формы данного состояния. Самая распространенная синфарингитная гематурия характеризуется резким началом на 2-3 сутки после инфекционного заболевания - фарингита, ларингита, кишечной инфекции. Иногда пусковым фактором болезни Берже могут выступать вакцинации, тяжелые физические нагрузки, длительные инсоляции (солнечный загар, посещение солярия). Пациенты жалуются на боль и дискомфортные ощущения в области поясницы, моча становится красноватого оттенка (макрогематурия), ее количество уменьшается. В редких случаях регистрируются признаки ОПН - задержка мочи с последующей полиурией, нарушения водно-солевого обмена. У некоторых пациентов приступ сопровождается повышением уровня артериального давления. Чаще всего при синфарингитной форме IgA-нефропатии почечная функция полностью восстанавливается через несколько дней или недель.
Латентная разновидность болезни характеризуется скрытым течением с практически полным отсутствием симптомов поражения почек. В анализе мочи определяются патологические изменения - выделение белка и небольших количеств крови. У ряда больных имеются жалобы на боли в мышцах, суставах, периодические отеки. Со временем при отсутствии лечения такая форма заболевания ведет к развитию хронической почечной недостаточности. Нефротическая форма, напротив, характеризуется выраженной клинической картиной - нефротическим синдромом с резкими отеками тела, развитием асцита, признаками обезвоживания по причине потери жидкости с мочой.
Осложнения
Основным и самым распространенным осложнением IgA-нефропатии является почечная недостаточность. Острые формы (ОПН) могут возникать при приступе синфарингитных разновидностей и иногда при нефротическом типе заболевания. Хроническая почечная недостаточность развивается медленно, регистрируется в течение 15 лет примерно у половины больных. Среди прочих осложнений нефропатии выделяют риски развития гиповолемического шока, нефротического криза, тромбозов на высоте приступа болезни. Отсутствие комплексного лечения во много раз повышает риск возникновения почечной недостаточности и других осложнений нефропатии.
Диагностика
В нефрологии определение патологии Берже производится на основании результатов общего осмотра пациента, сбора анамнеза, лабораторных данных (общих, биохимических и иммунологических показателей крови и мочи). Кроме того, в спорных случаях может быть назначено гистологическое исследование почек, цистоскопия, рентгенографические методы диагностики - в основном, для исключения других заболеваний. Осложняют диагностику патологии такие обстоятельства, как рецидивирующее течение синфарингитных форм (в период ремиссии проявления нефропатии практически не обнаруживаются) и отсутствие явной симптоматики при наличии латентных разновидностей. Все диагностические мероприятия при нефропатии разделяют на группы:
- Физикальный осмотр и сбор анамнеза. При расспросе пациента обращают внимание на частоту аллергических и инфекционных состояний в прошлом, пытаются найти их взаимосвязь с почечными проявлениями (гематурией, болью в пояснице). Нефротические типы нефропатии характеризуются наличием отеков, увеличением живота из-за асцита, симптомами почечной недостаточности.
- Лабораторные исследования крови. Изменения в общем анализе крови незначительные - при остром приступе возможно увеличение СОЭ, нерезко выраженный лейкоцитоз, повышение гематокрита. Биохимические показатели изменяются сильнее - увеличивается уровень глобулинов крови, креатинина (из-за нарушенной фильтрации в почках), при развитии нефротического синдрома возникает гипоальбуминемия и гиперлипидемия. Иммунологическое исследование крови указывает на рост уровня IgA и небольшое снижение фракций комплемента.
- Лабораторные исследования мочи. При синфарингитном типе патологии в моче отмечается макрогематурия, протеинурия до уровня 1-2 г/л, иммунологическое исследование обнаруживает наличие комплексов на основе IgA и незначительное количество компонентов комплемента (С3). Латентный вариант болезни Берже проявляется слабо выраженной протеинурией (до 0,3 г/л), наличием выщелоченных эритроцитов в моче.
- Инструментальная диагностика. УЗИ почек часто не выявляет специфических изменений на начальных этапах заболевания, лишь при длительном течении можно обнаружить незначительное уменьшение размеров органа. В основном УЗИ и УЗДГ почек применяются для дифференциальной диагностики. Экскреторная урография указывает на задержку контраста по причине пониженной фильтрационной способности.
- Гистологическое изучение.Биопсия почек с гистологическим исследованием материала является наиболее точным методом диагностики IgA-нефропатии. Обнаруживаются признаки воспаления в мезангиальном пространстве, гистохимическими методами в нем выявляются отложения иммунных комплексов.
Дифференциальную диагностику IgA-нефропатии проводят с другими формами нефропатии, мочекаменной болезнью, онкологическими поражениями органов мочевыделительной системы. Для этого больным назначают цистоскопию, рентгенологические и ультразвуковые исследования, определяют уровень уратов в биохимическом анализе крови. Вспомогательную роль в диагностике нефропатии играет выявление заболеваний, способных спровоцировать поражение почек - воспаления миндалин, гепатитов, кишечных инфекций.
Лечение болезни Берже
Этиотропное специфическое лечение не разработано, врачами-нефрологами используется симптоматическая, нефропротективная и поддерживающая терапия. При развитии почечной недостаточности показано назначение гемодиализа по показаниям. Трансплантация почек, применяющаяся в исключительных случаях, не является эффективным методом лечения - примерно у каждого второго больного в пересаженном органе развиваются аналогичные изменения. Это говорит о преимущественно экстраренальных причинах патологии. Наиболее часто при лечении заболевания используют следующие методы:
- Нефропротективная терапия. Применяют препараты, снижающие артериальное давление (ингибиторы АПФ, блокаторы ангиотензиновых рецепторов) и антиагреганты (дипиридамол). Помимо лекарственных средств для снижения нагрузки на выделительную систему рекомендуют поддержание оптимального водного режима, ограничение потребления поваренной соли.
- Антибактериальная терапия. Назначается в тех случаях, когда точно доказана взаимосвязь между нефропатией и наличием очага бактериальной инфекции (при синфарингитных формах). Выбор антибиотика и схема его приема зависят от характера возбудителя, что определяется при дополнительной диагностике. Иногда применяют метод удаления инфекционного очага (тонзиллэктомию).
- Противовоспалительная терапия. С этой целью назначают глюкокортикостероидные средства (преднизолон и его аналоги). Их используют при всех формах данной нефропатии, дозировка зависит от выраженности протеинурии, которая отражает степень повреждения почек.
- Иммуносупрессивная терапия. Применение цитостатиков и других иммуносупрессоров показано при тяжелых случаях и выраженном иммунологическом повреждении органов выделительной системы. Их включают в комплексную терапию нефротических форм болезни Берже.
В зависимости от наличия других симптомов повреждения почек и экстраренальных проявлений больным также показана инфузионная терапия, статины (для уменьшения уровня липидов крови), гипотензивные средства. Важен отказ от вредных привычек - курения, употребления алкоголя. Во избежание провоцирования приступа гематурии следует избегать физических нагрузок, длительного нахождения под солнцем.
Прогноз и профилактика
Прогноз болезни Берже неопределенный, зависит от показателей конкретного больного. По статистике, в течение 16-20 лет приблизительно у 30-50% пациентов развивается ХПН, но ее прогрессирование крайне медленное и доброкачественное. При правильном соблюдении предписаний врача и поддерживающей терапии качество жизни больных сохраняется на высоком уровне. Методы профилактики патологии не разработаны, некоторые специалисты рекомендуют своевременное полноценное лечение хронических инфекций (воспаления миндалин и других), но достоверных данных, что эти меры позволяют избежать синфарингитной нефропатии, нет. Профилактические мероприятия (водный режим, ограничение соли, исключение вредных привычек) дают возможность существенно замедлить прогрессирование повреждения почек.
Болезнь Гоше
Болезнь Гоше - это генетическое заболевание, характеризующееся нарушением липидного обмена, недостаточностью лизосомальных ферментов, накоплением гликолипидов в клеточных структурах. Симптомы определяются типом патологии. Общими признаками являются увеличение печени, селезенки, снижение свертываемости крови. При I типе выявляются нарушения со стороны костной системы: остеопороз, частые переломы, инфекции костей. При II и III типе доминирует неврологическая симптоматика: судороги, паралич, косоглазие, задержка умственного развития. Диагностика основана на биохимическом анализе дефицитарного фермента. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
МКБ-10
Заболевание получило свое название по фамилии французского врача Филиппа Гоше. В 1882 году он описал симптомы и патанатомические особенности строения селезенки пациентки, которая умерла от сепсиса. Спустя несколько десятилетий при аналогичном клиническом случае Гоше определил накопление в селезенке глюкоцереброзида и недостаточность фермента глюкоцереброзидазы. Болезнь Гоше (сфинголипидоз, глюкозилцерамидный липидоз) относится к группе лизосомальных болезней накопления - наследственных патологий, при которых изменены функции клеточных органелл лизосом. Частота заболевания составляет от 1:40 тыс. до 1:70 тыс. Распространенность наиболее велика в сообществах, где допустимы браки между близкими родственниками, например, у евреев ашкенази. Носительство мутационного гена определяется примерно у 1 человека из 400.
Глюкозилцерамидный сфинголипидоз является наиболее частой формой наследственных ферментопатий. Причиной его развития считается дефект гена GBA, который кодирует фермент лизосом бета-глюкозидазу (глюкоцереброзидазу), ответственную за расщепление липидов. Наследование болезни происходит аутосомно-рецессивным способом, для формирования ферментопатии необходимо присутствие пары измененных генов: один - от матери, другой - от отца. В супружеской паре, где оба родителя - носители мутации, вероятность рождения больного ребенка составляет 25%. Риск передачи одного дефектного гена, то есть риск носительства без развития болезни в таких семьях равен 50%. При наличии в генотипе двух мутантных аллелей функция глюкоцереброзидазы снижается на 15-30% от нормального уровня.
Патогенетической основой болезни является снижение каталитической активности бета-глюкозидазы. В результате нарушается процесс расщепления гликосфинголипидов (сложных соединений липидов и углеводов) до глюкозы и церамида. Аномально прогрессивное накопление макромолекул происходит в клетках, которые характеризуются повышенной скоростью их обновления - в макрофагах. Негидролизованные липиды концентрируются в лизосомах, образуются особые клетки накопления - клетки Гоше. Первичный метаболический сбой провоцирует вторичные расстройства биохимических процессов и клеточных функций. Из-за патологии жирового обмена развивается синдром активации макрофагов. Стимулируется моноцитопоэз, увеличивается содержание макрофагов в печени, селезенке, костном мозге. Это становится причиной спленомегалии, гепатомегалии, инфильтрации костного мозга. Расстройство регуляторной функции макрофагов является провоцирующим фактором цитопении, поражения костей и суставов.
Симптомы болезни Гоше
По возрасту дебюта и особенностям клинической картины выделяют три типа болезни. Первый тип наиболее распространен, имеет хронический характер течения. Симптомы чаще проявляются к 30-40 годам, реже болезнь манифестирует в детском возрасте. Увеличение размеров печени и селезенки начинается сразу после рождения, но клинически проявляется позже. Первыми признаками патологии становятся анемия, повышенная кровоточивость. Угнетение системы кроветворения сопровождается снижением уровня гемоглобина и тромбоцитов. Изменения со стороны опорно-двигательного аппарата представлены болями в костях и суставах, частыми переломами, деформациями (как правило, изменяется бедренная кость). У взрослых заметна гиперпигментация на лице и ногах: кожа темнеет, приобретает оттенок от желтоватого до желто-коричневого. Возможно появление плоских красных пятен с типичной локализацией в области вокруг глаз. Рост пациентов ниже среднего.
Второй тип болезни (острый инфантильный или острый нейропатический) встречается очень редко, развивается в промежутке от рождения до полутора лет, чаще всего симптомы дебютируют в первые три месяца жизни. Характеризуется стремительным течением, плохим откликом на лечение. На первый план выходят неврологические расстройства, спровоцированные скоплением клеток Гоше в центральной нервной системе. Дети слабо кричат, вяло сосут. Нарушен глотательный рефлекс, нередко отмечаются сбои цикла дыхания. Наблюдается заметная задержка психического и физического развития. На начальной стадии заболевания мышечный тонус снижен, через 9-12 месяцев после дебюта возникает гипертонус, особенно в мышцах шеи и конечностях. Развиваются судороги, косоглазие, спастический паралич. Печень и селезенка увеличены. Дети часто болеют тяжелой пневмонией.
Третий тип - ювенильный или подострый нейропатический. Первые признаки - увеличение селезенки и печени - возникают в 2-3 года. Полная симптоматика разворачивается в период с 6 до 15 лет. Клинические проявления поражения ЦНС включают гипертонус мышц, паралич спастического типа, косоглазие, непроизвольные спазмы, судороги, затрудненный цикл дыхания с трудностью вдоха, проблемы при глотании. Имеются расстройства психического развития: снижение интеллектуальных функций, несформированность речи и письма, эмоциональная неустойчивость, психозы. Дети отстают в половом развитии. Течение болезни неуклонно прогрессирующее.
Наиболее тяжелые осложнения выявляются при втором и третьем типе болезни. Поражение спинного и головного мозга приводит к нарушению дыхательного цикла, развиваются внезапные остановки дыхания, возрастает риск спазма гортани и смерти от удушья. Сниженный уровень тромбоцитов способен стать причиной обширных внутренних кровотечений. У больных с патологией первого типа распространенным осложнением является разрушение костей, их повышенная ломкость и инфекционные поражения. Ограничивается подвижность, пациенты не могут передвигаться самостоятельно, нуждаются в постороннем уходе.
Сбор анамнеза и физикальное обследование выполняется врачом-эндокринологом и неврологом, дополнительно назначаются консультации генетика, гематолога, офтальмолога, педиатра, психиатра. Анамнестические данные включают наличие болезни Гоше у родственников. При осмотре выявляются типичные признаки: низкий рост, патологии костей, неврологические симптомы (косоглазие, атаксия, паралич), геморрагический синдром, гиперпигментация кожи. Иногда подозрение на заболевание возникает после случайного выявления увеличенной селезенки на снимках УЗИ, угнетении кроветворной системы по данным общего анализа крови. Для подтверждения диагноза, исключения других метаболических наследственных патологий, остеомиелита, костного туберкулеза, вирусного гепатита и онкологических поражений крови проводится специфическая диагностика:
- Клиническое, биохимическое исследование крови. У большинства больных определяется тромбоцитопения, лейкопения, анемия, которая у детей обычно имеет железодефицитное происхождение. В результатах биохимического анализа обнаруживается сниженная активность глюкоцереброзидазы.
- Ферментный анализ клеток. При болезни Гоше в образцах сухой крови и в фибробластах кожи выявляется недостаточная активность глюкозидазы. Степень дефицитарности фермента не имеет прямой корреляции с выраженностью симптомов. Дополнительный биохимический маркер - хитотриозидаза. Этот фермент синтезируется активированными макрофагами, характерно повышение его активности в 6-10 раз.
- Морфологическое изучение костного мозга. Подтверждается наличие специфических для данного заболевания структур - клеток Гоше. Результат позволяет исключить гемобластоз и лимфопролиферативное заболевание.
- Исследование структуры костной ткани. С целью оценки тяжести поражения костно-суставной системы выполняется денситометрия, рентгенография и/или МРТ костей скелета. Возможен диффузный остеопороз, могут визуализироваться колбы Эрленмейера, очаги остеолизиса, остеосклероза и остеонекроза. На ранних стадиях болезни отмечается остеопения, инфильтрация костного мозга.
- Визуализирующее исследование селезенки, печени. Проводится УЗИ и МРТ внутренних органов. По результатам устанавливается наличие или отсутствие очаговых поражений, измеряется объем увеличенного органа. Исходные показатели в последующем позволяют контролировать эффективность терапии.
- Молекулярно-генетические исследования. ДНК-диагностика является необязательной процедурой. Подтверждение мутации в гене GBA бывает необходимо при неоднозначности биохимических исследований, а также в рамках пренатальных и преимплантационных обследований.
Лечение болезни Гоше
Специализированная помощь больным с первым и третьим типом болезни направлена на устранение симптомов и компенсацию первичного генетического дефекта - увеличение количества недостающего фермента, усиление катаболизма гликосфинголипидов. При 2 типе патологии терапевтические мероприятия оказываются недостаточно эффективными, усилия врачей сводятся к облегчению клинических проявлений - болей, судорог, дыхательных расстройств. Общая схема включает следующие направления:
- Ферментозаместительная терапия. Основным методом лечения является пожизненная ферментная заместительная терапия (ФЗТ) с применением рекомбинантной глюкоцереброзидазы. Эффективность достаточно высока - симптомы полностью купируются, качество жизни больных повышается. ФЗТ целесообразна при третьем и первом типе заболевания. Препараты вводятся внутривенно. Частые инфузии иногда становятся причиной воспалительных заболеваний вен (флебитов).
- Субстрат-редуцирующая терапия. Данное направление является новым в лечении болезни Гоше, относительно широко распространено в США и странах Европы. Нацелено на снижение скорости производства субстрата гликосфинголипидов и ускорение катаболизма накапливающихся макромолекул. В качестве препаратов выступают специфические ингибиторы глюкозилцерамидсинтазы. Метод показан при заболевании 1 типа с легкими и умеренными симптомами.
- Симптоматическая терапия. При явлениях остеопороза назначается комплексная терапия, включающая прием кальцийсодержащих препаратов, витамина D и соблюдение диеты, обогащенной кальцием. Эти меры позволяют замедлить потерю костной массы, повысить прочность костей, предотвратить переломы. При скелетных осложнениях применяются анальгезирующие средства (НПВС), антибактериальная терапия. Симптомы неврологических нарушений купируются противоэпилептическими препаратами, ноотропами, миорелаксантами.
Благоприятный исход наиболее вероятен у пациентов с 1 типом заболевания - комплексный терапевтический подход позволяет нормализовать функциональность глюкоцереброзидазы, предупредить развитие осложнений, избежать инвалидизации. При 3 типе прогноз зависит от характера течения болезни, индивидуальной реакции организма на лечебные мероприятия. 2 тип имеет крайне тяжелые проявления и завершается гибелью больного. Профилактика проводится во время планирования беременности и на ее начальных сроках. Медико-генетическое консультирование рекомендуется семьям, имеющим близких родственников с данной патологией. При высоком риске передачи мутации будущему ребенку в первом триместре выполняется исследование уровня фермента в амниотической жидкости, решается вопрос о прерывании беременности.
2. Болезнь Гоше: современная диагностика и лечение/ Лукина Е.А.// Клиническая онкогематология. - 2009 - Т.2, №2.
4. Болезнь Гоше: клиническая картина, диагностика, лечение/ Давыдкин И.Л., Хайретдинов Р.К., Кривова С.П., Данилова О.Е.// Эффективная фармакотерапия. - 2014 - №47.
Болезнь Гоше у детей. Клинические рекомендации.
Ферментная заместительная терапия - лечение, заключающееся в пожизненном введении препарата (рекомбинантного энзима) пациентам с врожденным дефектом метаболизма.
1. Краткая информация
1.1 Определение
Болезнь Гоше (БГ) - наиболее частая форма наследственных ферментопатий, объединенных в группу лизосомных болезней накопления, в основе которой лежит дефект гена GBA, кодирующего лизосомный фермент ?-D-глюкозидазу (глюкоцереброзидазу), ответственный за катаболизм липидов.
1.2 Этиология и патогенез
БГ наследуется по аутосомно-рецессивному типу. Присутствие двух мутантных аллелей гена GBA ассоциируется со значительным (? 30% от нормального уровня) снижением каталитической активности глюкоцереброзидазы, функция которой заключается в деградации гликосфинголипидов (или глюкоцереброзидов, глюкозилцерамидов) до глюкозы и церамидов. Дефицит фермента приводит к накоплению в лизосомах макрофагов неутилизированных липидов и образованию характерных клеток накопления (клеток Гоше). Следствием данного метаболического дефекта являются: хроническая активация макрофагальной системы, аутокринная стимуляция моноцитопоэза и увеличение абсолютного количества макрофагов, нарушение регуляторных функций макрофагов. Ген GBA, кодирующий глюкоцереброзидазу, расположен в хромосомной области 1q21. В настоящее время описано около 400 различных мутаций, патогенность которых проявляется широким полиморфизмом клинических симптомов, обусловленных частичной или полной потерей каталитической активности кодируемого фермента [1,2,3,4,5,6].
1.3 Эпидемиология
Частота БГ составляет 1:40000 - 1:70000. В популяции евреев-ашкенази (выходцев из Восточной Европы) частота встречаемости этого заболевания является более высокой и достигает 1:450 - 1:1000 [6].
1.4 Кодирование по МКБ-10
Е75.2 - Другие сфинголипидозы
1.5 Примеры диагнозов
- Болезнь Гоше, I тип.
- Болезнь Гоше, II тип.
- Болезнь Гоше, III тип.
- Болезнь Гоше, I тип. Состояние после спленэктомии.
- Болезнь Гоше, I тип. Патологический перелом шейки правого бедра, состояние после оперативного лечения.
- Болезнь Гоше, I тип. Асептический некроз головки бедренной кости слева.
- Болезнь Гоше, I тип. Остеопения поясничного отдела позвоночника.
- Болезнь Гоше, III тип. Симптоматическая эпилепсия.
- Болезнь Гоше, III тип. Косоглазие содружественное сходящееся альтернирующее.
1.6 Классификация
В зависимости от клинического течения выделяют 3 типа болезни Гоше:
- I тип - ненейронопатический (самый частый);
- II тип - инфантильный или острый нейронопатический;
- III тип - подострый нейронопатический.
При II и III типах БГ в патологический процесс вовлекается нервная система, поэтому их называют нейронопатическими [2,3,4,5,6].
2. Диагностика
2.1 Жалобы и анамнез
При сборе анамнеза и жалоб следует обратить внимание на наличие:
- задержки физического и полового развития;
- слабости, повышенной утомляемости, частых респираторных инфекций;
- проявлений спонтанного геморрагического синдрома (в виде подкожных гематом, кровоточивости слизистых оболочек) или длительных кровотечений при малых оперативных вмешательствах;
- болей в костях и суставах; давность, характер и локализацию болей, наличие в прошлом переломов костей;
- предшествующей спленэктомии (полной или частичной);
- неврологической симптоматики (глазодвигательная апраксия или сходящееся косоглазие, атаксия, потеря интеллекта, нарушения чувствительности и др.);
- семейного анамнеза (наличие спленэктомии или перечисленных выше симптомов у родных братьев и сестер [1,2,3,4,5,6]).
2.2 Физикальное обследование
- При проведении клинического осмотра рекомендуется включать измерение роста и массы тела, температуры тела, оценку состояния костно-суставной системы; выявление признаков геморрагического синдрома, гепатоспленомегалии, лимфаденопатии, а также своеобразной гиперпигментации кожных покровов в области коленных и локтевых суставов, характерной для пациентов с БГ [1,2,3,6].
Комментарии: Клинические проявления БГ I типа разнообразны, а возраст манифестации варьирует. БГ типа 1 имеет хроническое течение. Клиническая картина характеризуется прогрессирующим увеличением паренхиматозных органов (печени и селезенки), панцитопенией и патологией трубчатых костей скелета (болезненными костными кризами и аваскулярными некрозами эпифизов, чаще головки бедренной кости).
Основные клинические симптомы БГ I типа:
- гепатоспленомегалия,
- геморрагический синдром,
- костные боли (костные кризы),
- нарушение подвижности в суставах, обусловленное асептическим некрозом,
- патологические переломы,
- задержка физического и полового развития,
- астенический синдром.
Основные симптомы заболевания при БГ II типа возникают в первые 6 мес жизни (острая) нейропатическая форма с бульбарной и пирамидной симптоматикой, когнитивной задержкой. Течение заболевания - быстро прогрессирующее. Клинический симптомокомплекс включает признаки поражения нервной системы и внутренних органов:
- гепатоспленомегалия;
- нарушение глотания, поперхивание, часто осложняющиеся аспирационной пневмонией;
- тризм, билатеральное фиксированное косоглазие, прогрессирующая спастичность с ретракцией шеи, гиперрефлексия, положительный симптом Бабинского и другие патологические рефлексы;
- прогрессирующая задержка психомоторного развития и потеря ранее приобретенных навыков;
- тонико-клонические судорожные приступы, резистентные к противосудорожной терапии.
Главной особенностью клинических проявлений БГ III типа является то, что наряду с поражением паренхиматозных органов (гепатоспленомегалия) наблюдаются неврологические проявления, сходные с таковыми при типе 2 БГ, но менее тяжело выраженные и возникающие, как правило, в возрасте от 6 до 15 лет и позже:
- окуломоторные расстройства;
- снижение интеллекта (от незначительных изменений до тяжелой деменции);
- экстрапирамидная ригидность;
- мозжечковые нарушения;
- расстройства речи, письма;
- поведенческие изменения, эпизоды психоза;
- миоклонии, генерализованные тонико-клонические судороги.
В большинстве случаев течение заболевания медленно прогрессирующее [1,2,3,6].
2.3 Лабораторная диагностика
(Сила рекомендаций - 1; достоверность доказательств - С)
- Рекомендуется проведение клинического анализа крови [1,2,3,4,6].
Комментарии: у большинства больных с БГ выявляет тромбоцитопению, лейкопению и анемию, как проявления гиперспленизма.
- Рекомендовано морфологическое исследование костного мозга [1,2,3,4,6].
Комментарии: морфологическое исследование костного мозга способствует выявлению характерных диагностических элементов - клеток Гоше и одновременно исключению диагноза гемобластоза или лимфопролиферативного заболевания как причины цитопении и гепатоспленомегалии. Детям это исследование проводят редко, строго по показаниям.
- Рекомендуется проведение биохимического исследования: определение активности ?-D-глюкозидазы в лейкоцитах периферической крови, пятнах крови, высушенной на фильтровальной бумаге, определение активности хитотриозидазы в плазме крови [1,2,3,4,6].
- Рекомендовано проведение молекулярно-генетического исследования: выявление мутаций в гене GBА методом секвенирования кодирующих и прилегающих интронных областей [6].
- Рекомендуется оценить уровень сывороточного ферритина, ангиотензинпревращающего фермента, хемокина CCL 18 [6].
Комментарии. Характерными лабораторными симптомами при БГ также являются: повышение уровня сывороточного ферритина, ангиотензинпревращающего фермента, хемокина CCL 18, которые отражают степень активности заболевания и могут использоваться как биомаркеры для оценки динамики на фоне лечения.
2.4 Инструментальная диагностика
- Рекомендовано проведение рентгенографии костей скелета [1,2,3,4,6].
Комментарии: рентгенография костей скелета необходима для выявления и оценки тяжести поражения костно-суставной системы. Изменения костной ткани могут быть представлены диффузным остеопорозом, характерной колбообразной деформацией дистальных отделов бедренных и проксимальных отделов большеберцовых костей (колбы Эрленмейера), очагами остеолизиса, остеосклероза и остеонекроза, патологическими переломами.
- Рекомендовано проведение денситометрии и магнитно-резонансной томографии (МРТ) [1,2,3,4,6].
Комментарии: денситометрия и магнитно-резонансная томография (МРТ являются более чувствительными методами и позволяют диагностировать поражение костей (остеопению, инфильтрацию костного мозга) на ранних стадиях, не доступных визуализации рентгенографией.
- Рекомендовано проведение ультразвукового исследования (УЗИ) и МРТ печени и селезенки [1,2,3,4,6].
Комментарии: УЗИ и МРТ печени и селезенки позволяют выявить их очаговые поражения и определить исходный объем органов, что необходимо для последующего контроля эффективности заместительной ферментной терапии.
- Рекомендовано проведение допплер-эхокардиографии у спленэктомированных пациентов [3,5,6].
- Рекомендовано проведение эзофагогастродуоденоскопия при наличии соответствующих жалоб или признаков портальной гипертензии [3,5,6].
2.4 Иная диагностика
Консультации специалистов пациентам с подозрением на болезнь Гоше рекомендуются по показаниям.
- Рекомендована консультации психоневролога [1,2,3,4,5,6]
Комментарии: необходима всем детям с БГ для уточнения типа заболевания.
Комментарии: показана при подозрении на наличие у ребенка скелетной патологии.
- Рекомендована консультация врача-генетика [1,2,3,4,5,6].
Комментарии: показана семьям, имеющим родственников с болезнью Гоше.
2.5. Медико-генетическое консультирование и пренатальная диагностика
- Семьям с больными детьми рекомендуется медико-генетическое консультирование с целью разъяснения генетического риска [1,2,3,4,5,6].
Комментарии: как и при других аутосомно-рецессивных заболеваниях, при БГ для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, существует возможность проведения пренатальной и преимплантационной диагностики.
Пренатальная диагностика проводится молекулярно-генетическими или биохимическими методами, путем исследования ДНК, выделенной из биоптата ворсин хориона на 9-11-й неделе беременности и/или клеток амниотической жидкости, плодной крови на 20-22-й неделе беременности.
2.6 Дифференциальная диагностика
Диагноз болезнь Гоше ставится на основании совокупности клинических данных, результатов лабораторного исследования, биохимического и молекулярно-генетического анализа.
Диагноз БГ следует заподозрить у пациента с необъяснимой сплено- и гепатомегалией, цитопенией и симптомами поражения костей.
Золотым стандартом диагностики является биохимическое тестирование, поскольку патогенность некоторых выявленных редких и новых мутаций требует дополнительных доказательств [2,6].
Для I типа БГ в зависимости от вида манифестации - разнообразные экзогенные и наследственные заболевания, сопровождающиеся висцеромегалией, острыми болями в костях, кровоточивостью (вирусные гепатиты, остеомиелит, костный туберкулез, гемофилия, гликогеноз, болезнь Нимана-Пика (тип В), недостаточность кислой липазы).
Для II и III типов БГ - болезнь Нимана-Пика (типы А, С), GM1-ганглиозидоз, галактосиалидоз, дефицит лизосомной кислой липазы, а также врожденная окуломоторная апраксия.
3. Лечение
3.1 Консервативное лечение
- Рекомендована пожизненная ферментная заместительная терапия (ФЗТ) рекомбинантной глюкоцереброзидазой пациентам с подтвержденным диагнозом БГ I типа без поражения нервной системы [1,2,3,5,6].
(Сила рекомендаций - 1; достоверность доказательств - В)
- Рекомендована пожизненная ферментная заместительная терапия (ФЗТ) рекомбинантной глюкоцереброзидазой пациентам с хроническим поражением нервной системы (БГ III тип), у которых имеются клинически значимые неневрологические проявления заболевания [2,4,5,6].
(Сила рекомендаций - 2; достоверность доказательств - В)
Комментарии: ФЗТ обеспечивает устойчивое улучшение состояния пациентов: нормализует уровни гемоглобина, тромбоцитов; объем печени и селезенки (у неспленэктомизированных больных); купирует костные боли, предотвращает развитие остеонекрозов и костных кризов; приводит к нормализации роста и значительно повышает качество жизни детей с болезнью Гоше.
- имиглюцераза ж,7н ;
- велаглюцераза альфа ж,7н .
Имиглюцераза ж,7н - модифицированная форма кислой ?-глюкозидазы человека, у детей с БГ начальная доза имиглюцеразы составляет:
- при I типе БГ без поражения трубчатых костей скелета - 30 ЕД/кг на 1 введение [6,7,8];
- при I типе БГ с поражением трубчатых костей скелета (костные кризы, патологические переломы, очаги литической деструкции, асептический некроз головок бедренных костей) - 60 ЕД/кг на 1 введение [6,7,8,9,10,11];
- при III типе БГ - 60 ЕД/кг на 1 введение.
Велаглюцераза альфа ж,7н показана для длительного лечения детей с болезнью Гоше I типа. Рекомендуемая доза составляет 30-60 ЕД/кг 1 раз в 2 недели.
Дозу можно корректировать индивидуально, на основании достижения ожидаемого эффекта и его сохранения, однако применение доз выше 60 ЕД/кг не изучено [6,12,13,14,15].
При обследовании сиблингов (братьев и сестер пробанда) могут быть выявлены дети с БГ, не имеющие клинических проявлений. Такие пациенты нуждаются в наблюдении, начинать их лечение необходимо при появлении первых симптомов болезни.
- Не рекомендована ферментная заместительная терапия при II типе БГ, так как не эффективна [1,2,3,5,6].
(Сила рекомендаций - 2; достоверность доказательств - С)
- При развитии проявлений остеопороза для замедления и прекращения потери костной массы, повышения ее прочности, предотвращения переломов костей в комплексной терапии рекомендуется применять: альфакальцидол ж,вк , соли кальция ж,вк [1,3,5,6]
(Сила рекомендаций - 1; достоверность доказательств - С)
Комментарии: в качестве симптоматической терапии скелетных осложнений БГ I типа назначаются анальгетики (во время костных кризов), нестероидные противовоспалительные средства, редко, лишь при наличии показаний, - антибактериальная терапия.
- Не рекомендовано проведение спленэктомии [1,3,5,6].
- При доказанном диагнозе БГ не рекомендованы повторные пункции костного мозга и другие инвазивные диагностические мероприятия (биопсия печени, селезенки) [1,3,5,6].
- Не рекомендовано оперативное лечение костных кризов, которые ошибочно рассматриваются как проявления остеомиелита. При хирургических вмешательствах существует повышенный риск кровотечения и инфицирования [1,2,5,6].
- Противопоказано назначение кортикостероидов с целью купирования цитопенического синдрома [1,2,5,6].
- Не рекомендовано и не обосновано назначение препаратов железа больным с развернутой картиной БГ, так как анемия в этих случаях носит характер «анемии воспаления» [1,2,5,6].
Болезнь Гоше у детей
Болезнь Гоше (БГ) - наиболее частая форма наследственных ферментопатий, объединенных в группу лизосомных болезней накопления, в основе которой лежит дефект гена GBA, кодирующего лизосомный фермент β-D-глюкозидазу (глюкоцереброзидазу), ответственный за катаболизм липидов.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
• Болезнь Гоше, I тип. Патологический перелом шейки правого бедра, состояние после оперативного лечения.
Этиология и патогенез
БГ наследуется по аутосомно-рецессивному типу. Присутствие двух мутантных аллелей гена GBA ассоциируется со значительным ( ≤ 30% от нормального уровня) снижением каталитической активности глюкоцереброзидазы, функция которой заключается в деградации гликосфинголипидов (или глюкоцереброзидов, глюкозилцерамидов) до глюкозы и церамидов.
Дефицит фермента приводит к накоплению в лизосомах макрофагов неутилизированных липидов и образованию характерных клеток накопления (клеток Гоше).
Следствием данного метаболического дефекта являются: хроническая активация макрофагальной системы, аутокринная стимуляция моноцитопоэза и увеличение абсолютного количества макрофагов, нарушение регуляторных функций макрофагов. Ген GBA, кодирующий глюкоцереброзидазу, расположен в хромосомной области 1q21. В настоящее время описано около 400 различных мутаций, патогенность которых проявляется широким полиморфизмом клинических симптомов, обусловленных частичной или полной потерей каталитической активности кодируемого фермента [1,2,3,4,5,6].
Эпидемиология
• проявлений спонтанного геморрагического синдрома (в виде подкожных гематом, кровоточивости слизистых оболочек) или длительных кровотечений при малых оперативных вмешательствах;
• болей в костях и суставах; давность, характер и локализацию болей, наличие в прошлом переломов костей;
• неврологической симптоматики (глазодвигательная апраксия или сходящееся косоглазие, атаксия, потеря интеллекта, нарушения чувствительности и др.);
• семейного анамнеза (наличие спленэктомии или перечисленных выше симптомов у родных братьев и сестер [1,2,3,4,5,6]).
• При проведении клинического осмотра рекомендуется включать измерение роста и массы тела, температуры тела, оценку состояния костно-суставной системы; выявление признаков геморрагического синдрома, гепатоспленомегалии, лимфаденопатии, а также своеобразной гиперпигментации кожных покровов в области коленных и локтевых суставов, характерной для пациентов с БГ [1,2,3,6].
Комментарии: Клинические проявления БГ I типа разнообразны, а возраст манифестации варьирует. БГ типа 1 имеет хроническое течение. Клиническая картина характеризуется прогрессирующим увеличением паренхиматозных органов (печени и селезенки), панцитопенией и патологией трубчатых костей скелета (болезненными костными кризами и аваскулярными некрозами эпифизов, чаще головки бедренной кости).
Основные симптомы заболевания при БГ II типа возникают в первые 6 мес жизни (острая) нейропатическая форма с бульбарной и пирамидной симптоматикой, когнитивной задержкой. Течение заболевания - быстро прогрессирующее. Клинический симптомокомплекс включает признаки поражения нервной системы и внутренних органов:
• тризм, билатеральное фиксированное косоглазие, прогрессирующая спастичность с ретракцией шеи, гиперрефлексия, положительный симптом Бабинского и другие патологические рефлексы;
Главной особенностью клинических проявлений БГ III типа является то, что наряду с поражением паренхиматозных органов (гепатоспленомегалия) наблюдаются неврологические проявления, сходные с таковыми при типе 2 БГ, но менее тяжело выраженные и возникающие, как правило, в возрасте от 6 до 15 лет и позже:
Комментарии: у большинства больных с БГ выявляет тромбоцитопению, лейкопению и анемию, как проявления гиперспленизма.
• Рекомендуется проведение биохимического исследования: определение активности β-D-глюкозидазы в лейкоцитах периферической крови, пятнах крови, высушенной на фильтровальной бумаге, определение активности хитотриозидазы в плазме крови [1,2,3,4,6].
• Рекомендовано проведение молекулярно-генетического исследования: выявление мутаций в гене GBА методом секвенирования кодирующих и прилегающих интронных областей [6].
• Рекомендуется оценить уровень сывороточного ферритина, ангиотензинпревращающего фермента, хемокина CCL 18 [6].
Комментарии. Характерными лабораторными симптомами при БГ также являются: повышение уровня сывороточного ферритина, ангиотензинпревращающего фермента, хемокина CCL 18, которые отражают степень активности заболевания и могут использоваться как биомаркеры для оценки динамики на фоне лечения.
• Рекомендовано проведение эзофагогастродуоденоскопия при наличии соответствующих жалоб или признаков портальной гипертензии [3,5,6].
• Семьям с больными детьми рекомендуется медико-генетическое консультирование с целью разъяснения генетического риска [1,2,3,4,5,6].
Комментарии: как и при других аутосомно-рецессивных заболеваниях, при БГ для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, существует возможность проведения пренатальной и преимплантационной диагностики.
Пренатальная диагностика проводится молекулярно-генетическими или биохимическими методами, путем исследования ДНК, выделенной из биоптата ворсин хориона на 9-11-й неделе беременности и/или клеток амниотической жидкости, плодной крови на 20-22-й неделе беременности.
Дифференциальный диагноз
Диагноз болезнь Гоше ставится на основании совокупности клинических данных, результатов лабораторного исследования, биохимического и молекулярно-генетического анализа.
Диагноз БГ следует заподозрить у пациента с необъяснимой сплено- и гепатомегалией, цитопенией и симптомами поражения костей.
Золотым стандартом диагностики является биохимическое тестирование, поскольку патогенность некоторых выявленных редких и новых мутаций требует дополнительных доказательств [2,6].
Для I типа БГ в зависимости от вида манифестации - разнообразные экзогенные и наследственные заболевания, сопровождающиеся висцеромегалией, острыми болями в костях, кровоточивостью (вирусные гепатиты, остеомиелит, костный туберкулез, гемофилия, гликогеноз, болезнь Нимана-Пика (тип В), недостаточность кислой липазы).
Для II и III типов БГ - болезнь Нимана-Пика (типы А, С), GM1-ганглиозидоз, галактосиалидоз, дефицит лизосомной кислой липазы, а также врожденная окуломоторная апраксия.
Лечение
• Рекомендована пожизненная ферментная заместительная терапия (ФЗТ) рекомбинантной глюкоцереброзидазой пациентам с подтвержденным диагнозом БГ I
• Рекомендована пожизненная ферментная заместительная терапия (ФЗТ) рекомбинантной глюкоцереброзидазой пациентам с хроническим поражением нервной системы (БГ III тип), у которых имеются клинически значимые неневрологические проявления заболевания [2,4,5,6].
• Не рекомендована ферментная заместительная терапия при II типе БГ, так как не эффективна [1,2,3,5,6].
Комментарии: ФЗТ обеспечивает устойчивое улучшение состояния пациентов: нормализует уровни гемоглобина, тромбоцитов; объем печени и селезенки (у неспленэктомизированных больных); купирует костные боли, предотвращает развитие остеонекрозов и костных кризов; приводит к нормализации роста и значительно повышает качество жизни детей с болезнью Гоше.
- велаглюцераза альфа ж,7н .
Имиглюцераза ж,7н - модифицированная форма кислой β-глюкозидазы человека, у детей с БГ начальная доза имиглюцеразы составляет:
- при I типе БГ без поражения трубчатых костей скелета - 30 ЕД/кг на 1 введение [6,7,8];
Велаглюцераза альфа ж,7н показана для длительного лечения детей с болезнью Гоше I типа. Рекомендуемая доза составляет 30-60 ЕД/кг 1 раз в 2 недели.
Дозу можно корректировать индивидуально, на основании достижения ожидаемого эффекта и его сохранения, однако применение доз выше 60 ЕД/кг не изучено [6,12,13,14,15].
При обследовании сиблингов (братьев и сестер пробанда) могут быть выявлены дети с БГ, не имеющие клинических проявлений. Такие пациенты нуждаются в наблюдении, начинать их лечение необходимо при появлении первых симптомов болезни.
• При развитии проявлений остеопороза для замедления и прекращения потери костной массы, повышения ее прочности, предотвращения переломов костей в комплексной терапии рекомендуется применять: альфакальцидолж,вк, соли кальцияж,вк [1,3,5,6]
Комментарии: в качестве симптоматической терапии скелетных осложнений БГ I типа назначаются анальгетики (во время костных кризов), нестероидные противовоспалительные средства, редко, лишь при наличии показаний, - антибактериальная терапия.
• При доказанном диагнозе БГ не рекомендованы повторные пункции костного мозга и другие инвазивные диагностические мероприятия (биопсия печени, селезенки) [1,3,5,6].
• Не рекомендовано оперативное лечение костных кризов, которые ошибочно рассматриваются как проявления остеомиелита. При хирургических вмешательствах существует повышенный риск кровотечения и инфицирования [1,2,5,6].
• Противопоказано назначение кортикостероидов с целью купирования цитопенического синдрома [1,2,5,6].
• Не рекомендовано и не обосновано назначение препаратов железа больным с развернутой картиной БГ, так как анемия в этих случаях носит характер «анемии воспаления» [1,2,5,6].
Ведение пациентов рекомендуется осуществлять в соответствии с рекомендациями по минимально необходимому мониторингу состояния больных при БГ I типа, разработанными Объединенной международной группой по изучению болезни Гоше (International Collaborative Gaucher Group). Контроль показателей крови необходимо проводить 1 раз в 3 мес, размеров паренхиматозных органов (УЗИ, МРТ) - 1 раз в 6 мес, а также при изменении дозировки препарата или при значительных клинических осложнениях (табл. 1). Определение состояния костной ткани осуществляют 1 раз в год. Определение активности хитотриазидазы на фоне ферментной заместительной терапии проводят 1 раз в 12 мес.
Рекомендуется пациентов с БГ наблюдать по месту жительства в амбулаторно-поликлинических условиях врачам педиатрам, гематологам, при БГ III типа - неврологам, при наличии костных нарушений - ортопедам, до достижения возраста 18 лет.
Рекомендуется введение ФЗТ проводить регулярно при наличии показаний в случае осложненного течения болезни - в условиях круглосуточного стационара, в стабильном состоянии - в стационаре дневного пребывания или амбулаторно 1 раз в 2 недели. До достижения клинико-лабораторной ремиссии все пациенты с БГ должны проходить контрольное обследование с целью коррекции дозы ФЗТ в круглосуточном или дневном стационаре 2 раза в год; в дальнейшем обследование проводится 1 раз в год.
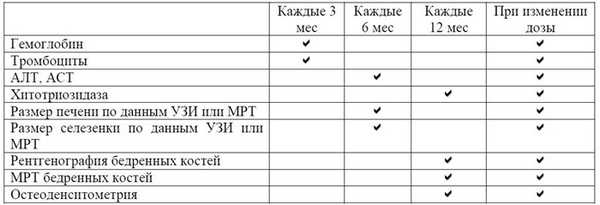
Таблица 1- Принципы мониторинга болезни Гоше
АЛТ/АСТ ― аланин-/аспартатаминотрансферазы, УЗИ ― ультразвуковое исследование, МРТ ― магнитно-резонансная томография.
Читайте также: