Синдром Фишера-Бушке (Fischer-Buschke) - синонимы, авторы, клиника
Добавил пользователь Валентин П. Обновлено: 01.02.2026
наследственная болезнь, проявляющаяся в раннем детском возрасте сочетанием кератодермии, ладонно-подошвенного гипергидроза, онихогрифоза, переходящего в онихолизис, скудного роста волос и утолщения дистальных фаланг пальцев кистей и стоп; наследуется по аутосомно-доминантному типу.
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
Смотреть что такое "Фишера синдром" в других словарях:
Фишера синдром — (Н. Fischer, 1884 1943, нем. дерматолог; син. Бушке Фишера синдром) наследственная болезнь, проявляющаяся в раннем детском возрасте сочетанием кератодермии, ладонно подошвенного гипергидроза, онихогрифоза, переходящего в онихолизис, скудного… … Большой медицинский словарь
ФИШЕРА СИНДРОМ — - см. Миллера Фишера синдром … Энциклопедический словарь по психологии и педагогике
Бушке-Фишера синдром — (A. Buschke, Н. Fischer) см. Фишера синдром … Большой медицинский словарь
МИЛЛЕРА ФИШЕРА СИНДРОМ — - эпонимическое название двух синдромов, описанных канадским неврологом С. M. Fisher (род. в 1913). 1. Вариант синдрома Гийена - Барре, проявляющийся арефлексией, атаксией (мозжечкового характера) и офтальмоплегией (с вовлечением наружных, реже… … Энциклопедический словарь по психологии и педагогике
Экономо-Фишера синдром — (К. Economo, 1876 1931, австрийский невропатолог; Fischer) гемибаллизм, наблюдаемый при нарушениях кровообращения в головном мозге с поражением подбугорного ядра (люисова тела) … Большой медицинский словарь
Синдром Гийена — У этой статьи нет иллюстраций. Вы можете помочь проекту, добавив их (с соблюдением правил использования изображений). Для поиска иллюстраций можно: попробовать воспользоваться инструментом … Википедия
синдром периоральный — (syndromum periorale; греч. peri вокруг, около + лат. os, oris рот) см. Фишера Брюгге синдром … Большой медицинский словарь
Фишера-Брюгге синдром — (Н. Fischer, 1884 1943, нем. дерматолог; Brugge; син. синдром периоральный) стойкое покраснение кожи вокруг рта; проявление ангиотрофоневроза … Большой медицинский словарь
Фишера-Эванса синдром — (J. A. Fisher; R. S. Evans, совр. амер. врач) см. Эванса синдром … Большой медицинский словарь
ФИШЕРА - ЭВАНСА СИНДРОМ — - см. Эванса синдром … Энциклопедический словарь по психологии и педагогике
ЭВАНСА СИНДРОМ — (синдром Фишера - Эванса, по именам американских врачей R. S. Evans, род. в 1912, и J. A. Fisher) - сочетание иммунной тромбоцитопении и Кумбс положительной гемолитической анемии. Может быть проявлением антифосфолипидного синдрома. Лечение:… … Энциклопедический словарь по психологии и педагогике
Синдром Фишера-Бушке (Fischer-Buschke) - синонимы, авторы, клиника
ГБОУ ВПО «Сургутский государственный университет ХМАО-Югры», Сургут, Россия, 628403
БУ ХМАО-Югры «Сургутский клинический кожно-венерологический диспансер», Сургут, Россия, 628403
Случай кератодермии Бушке-Фишера, ассоциированной с ревматоидным артритом
Журнал: Клиническая дерматология и венерология. 2016;15(1): 15‑17
В статье представлено клиническое наблюдение кератодермии Бушке—Фишера в сочетании с ревматоидным артритом.
Дерматозы с преимущественной локализацией в области ладоней и подошв, протекающие с явлениями гиперкератоза кожи, включают обширную группу заболеваний и представляют определенный интерес для специалистов ввиду частоты встречаемости, сложностей дифференциальной диагностики, а также возникающих трудностей в лечении [1].
Кератодермии — одна из наиболее распространенных групп генодерматозов. После ихтиозиформных поражений кожи данная группа заболеваний занимает второе место в структуре наследственных болезней кожи [2—4].
Диссеминированная пятнистая кератодермия Бушке—Фишера (син.:keratodermia maculosa disseminata Buschke—Fischer, наследственная ладонно-подошвенная рассеянная кератома, рассеянный точечный кератоз Бушке—Фишера) наследуется аутосомно-доминантно. Доказано повреждение в гене AAGAB, кодирующем α- и γ-адаптинсвязывающий белок [2].
Заболевание проявляется чаще в молодом возрасте (15—30 лет). На коже ладоней, подошв, пальцев возникают мелкие внутрироговые «жемчужины», превращающиеся в роговые, плотные, коричневые пробки диаметром до 1 см с кратерообразными краями. Очаги поражения не сливаются. После отторжения роговых пробок остаются кратерообразные углубления с роговыми стенками. Иногда они покрываются твердыми корками и становятся бугристыми. Потоотделение не нарушено [4—7].
У большинства больных процесс асимптомный, однако при большом количестве элементов возможны значительная болезненность и затруднения при ходьбе [3].
Клинический случай
Пациентка Р., 35 лет, жительница города Сургута, обратилась к дерматовенерологу Сургутского клинического кожно-венерологического диспансера с жалобами на высыпания на коже верхних и нижних конечностей без субъективных ощущений.
Анамнез заболевания: считает себя больной с 15 лет, когда впервые появились высыпания на коже ладоней и стоп. Дебют заболевания произошел на фоне перенесенной неспецифической инфекции (названия не помнит) и последующего развития ревматоидного артрита. За медицинской помощью по поводу кожных проявлений не обращалась, считая высыпания на коже кистей, стоп «семейной особенностью». Периодически (раз в несколько месяцев) срезала кожные образования при помощи ножниц.
Наследственность отягощена — подобные высыпания имелись у матери, родной сестры, у деда по материнской линии, у которого, со слов пациентки, также были высыпания на ладонях и стопах, которые он периодически состригал.
Анамнез жизни: туберкулез, описторхоз, вирусный гепатит отрицает. Из хронических заболеваний отмечает ревматоидный артрит, наблюдается у терапевта и ревматолога, принимает метотрексат в дозе 5 мг в неделю. Травмы в прошлом отрицает. Аллергологический анамнез: непереносимость пенициллина (реакция в виде сыпи).
Объективно: общее состояние удовлетворительное, кожные покровы и видимые слизистые оболочки физиологической окраски, периферические лимфатические узлы не увеличены. Со стороны костно-мышечного аппарата отмечаются деформация пястно-фаланговых суставов и отклонение I—IV пальцев стоп в латеральную сторону («плавник моржа»). Дыхание везикулярное, хрипов нет. Тоны сердца ритмичные, ясные. Артериальное давление 120/80 мм рт.ст. Живот при пальпации мягкий, безболезненный во всех отделах. Печень по краю реберной дуги, селезенка не увеличена. Стул и диурез без особенностей.
Патологический кожный процесс локализуется симметрично на коже ладоней и подошвенной поверхности стоп. Представлен в виде равномерно рассеянных плоских очажков ороговения желтоватого цвета округлых очертаний, размерами от 0,3 до 1,0 см (рис. 1, 2). На подошвенной поверхности стоп папулы сливаются в очаги размером от 1,5 до 2,5 см. При удалении роговых масс остается кратерообразное углубление. Потоотделение не нарушено. Ногтевые пластины кистей и стоп не изменены. Других патологических высыпаний не наблюдается.
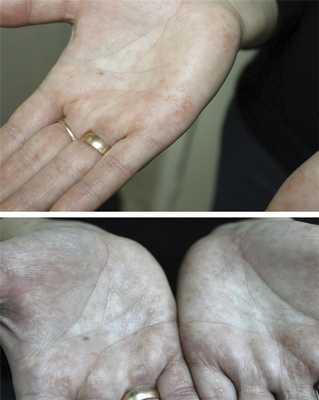
Рис. 1. Поражение ладоней у пациентки с кератодермией Бушке—Фишера.
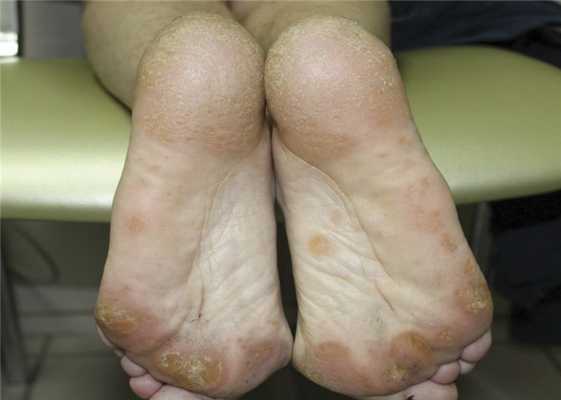
Рис. 2. Поражение подошвенных поверхностей стоп у пациентки с кератодермией Бушке—Фишера.
Данные лабораторных исследований. Гемограмма (от 17.10.14): лейкоциты 12,6·10 9 /л, эритроциты 4,55·10¹²/л, тромбоциты 492·10 9 /л, гемоглобин 106 г/л, СОЭ 26 мм/ч, палочкоядерные 0%, сегментоядерные 87,9%, эозинофилы 0,3%, моноциты 2,5%, лимфоциты 9%, базофилы 0,3%.
Общий анализ мочи (от 17.10.14): плотность 1015, белок, эритроциты, глюкоза отрицательно, эпителий единичный в поле зрения, лейкоциты 0—3 в поле зрения, кетоновые тела, соли отрицательно.
Биохимия крови (от 17.10.14): аланинаминотрансфераза 35 ЕД/л, аспартатаминотрансфераза 37,4 ЕД/л, креатинин 42,9 мкмоль/л, холестерин 4,1 ммоль/л, общий белок 83,5 г/л, щелочная фосфатаза 291,01 ЕД/л, общий билирубин 10,64 мкмоль/л, глюкоза 5,70 ммоль/л.
Реакция микропреципитации (от 22.09.14) отрицательная.
Микроскопия чешуек стоп, ногтей стоп на патогенные грибы (от 20.10.14): элементы дерматомицетов не обнаружены.
Бакпосев на дерматомицеты (от 20.10.14) чешуек стоп — роста нет.
На основании данных анамнеза (дебют заболевания в 15 лет, семейный характер высыпаний: у деда по материнской линии, матери, родной сестры), характерных клинических проявлений (симметрично расположенные папулы желтого цвета на коже ладоней и подошв, отсутствие нарушений потоотделения) выставлен диагноз «наследственная кератодермия Бушке—Фишера». Сопутствующий диагноз «ревматоидный артрит, поздняя стадия, средней активности» (заключение ревматолога).
Пациентке рекомендованы терапия ревматоидного артрита, диспансерное наблюдение дерматовенеролога в Сургутском клиническом кожно-венерологическом диспансере, курсовой прием витаминов, А и Е (аевит по 1 капсуле 1 раз в день в течение 1 мес с перерывом в 1 мес, всего 5 курсов в год) [8], уход за кожей кистей, стоп (мыльно-содовые ванночки регулярно 1 раз в неделю, механическая чистка роговых наслоений кожи стоп либо применение раствора Аквапилинг, использование эмолентов: крем Локобейз-рипеа 1—2 раза в день на кожу стоп), ношение мягкой, не травмирующей обуви, санаторно-курортное лечение (Пятигорск, Саки, Евпатория, Серноводск).
Заключение
Наследственные кератодермии, несмотря на относительную изученность, сравнительно редко встречаются в практике врача-дерматовенеролога и, как следствие, вызывают затруднения при диагностике. Данное наблюдение интересно сочетанием двух заболеваний, одно из которых представляет собой генетически детерминированное нарушение процесса ороговения.
Российский государственный медицинский университет им. Н.И. Пирогова
ФГБОУ ВО «Российский национальный исследовательский университет им. Н.И. Пирогова», Москва, Россия;
Российская детская клиническая больница, Москва, Россия
Сложный диагностический случай: синдром Бушке—Оллендорф или соединительнотканный невус?
Дисплазия соединительной ткани, представляющая собой генетически детерминированное нарушение закладки и постнатального развития, является весомой проблемой современной медицины в силу широкого распространения и возможных тяжелых последствий. Клинические проявления дисплазии соединительной ткани варьируют в широких пределах — от повышенной растяжимости кожи до сосудистых аномалий, приводящих к внезапной смерти. Большую группу в пределах дисплазии соединительной ткани составляют различные невусы, изолированные или в составе различных синдромов. Одним из редких генетически детерминированных синдромов является сидром Бушке—Оллендорф, относящийся к орфанным заболеваниям с аутосомно-доминантным наследованием. Основные клинические проявления синдрома — распространенные соединительнотканные невусы кожи, выявляемые в раннем детском возрасте, и остеопойкилия, чаще диагностируемая у взрослых. К редким проявлениям синдрома Бушке—Оллендорф относят различные пороки и поражения нервной системы (от нарушения когнитивного развития до эпилепсии). Диагностика синдрома основывается на совокупности определенных симптомов, главными из которых являются соединительнотканные невусы и остеопойкилия. В случае неполного синдрома диагностика базируется на молекулярно-генетическом исследовании, которое в силу дороговизны доступно не всем пациентам. Данный клинический случай демонстрирует трудность диагностического поиска и неоднозначность получаемых при этом результатов. Представленный клинический случай был обсужден на общегородском Московском консилиуме при участии дерматовенерологов Москвы и профессоров Н.Н. Потекаева, В.Г. Акимова, В.Н. Гребенюка, А.Н. Львова, Э.А. Баткаева, Н.Г. Короткого, О.В. Жуковой, В.А. Волнухина.
Дисплазия соединительной ткани — это нарушение развития соединительной ткани в эмбриональном и постнатальном периодах, генетически детерминированное, характеризующееся дефектами волокнистых структур и основного вещества соединительной ткани, приводящее к расстройству гомеостаза на тканевом, органном и организменном уровнях в виде различных морфофункциональных нарушений висцеральных и локомоторных органов с прогредиентным течением, определяющее особенности ассоциированной патологии. По самым скромным данным, показатели распространенности данной группы заболеваний соотносятся с распространенностью основных социально значимых неинфекционных заболеваний. К данной группе заболеваний относится диссеминированный лентикулярный дерматофиброз (синдром Бушке—Оллендорф), являющийся редким наследственным заболеванием [1—3]. Впервые заболевание было описано Абрахамом Бушке в 1902 г. под названием «scleroderma adultorum». В 1915 г. Генрих Эрнст Альберс-Шёнберг, а затем в 1928 г. Элен Оллендорф описали еще 2 случая заболевания.
Синдром Бушке—Оллендорф представляет собой сочетание соединительнотканных невусов кожи и остеопойкилии (OMIM 166700). Частота встречаемости данного заболевания составляет 1 на 20—30 тыс. живых новорожденных, с одинаковой частотой у мальчиков и девочек. Заболевание наследуется аутосомно-доминантно с высокой пенетрантностью и вариабельной экспрессивностью. В основе лежит мутация гена LEMD3, расположенного на 12q14.3 хромосоме, кодирующего один из структурных белков соединительной ткани, что сопровождается чрезмерным накоплением эластина в дерме [4].
Синдром Бушке—Оллендорф обычно проявляется рано, возможна манифестация первых симптомов сразу после рождения. Как и при любом пороке развития, симптоматика весьма разнообразна и подразделяется на кожные и внекожные признаки (рис. 1).

Рис. 1. Клинические проявления синдрома Бушке—Оллендорф.
Первым симптомом заболевания является поражение кожного покрова в виде соединительнотканного невуса. Высыпания обычно локализуются на боковых поверхностях туловища, верхней трети живота, спины, пояснице, бедрах, ягодицах; представлены папулами, узлами и бляшками, слегка возвышающимися над уровнем кожи, телесного или желтоватого цвета, склонными к сетевидной, линейной или герпетиформной ориентации; могут сливаться между собой. Элементы сыпи, как правило, симметричны, однако в детском возрасте описано одностороннее расположение. Консистенция элементов мягкая, пальпация безболезненна. Субъективных ощущений от эффлоресценций обычно нет. Из других кожных изменений описаны анетодермия, ладонно-подошвенный кератоз, морфея, околосуставные подушечки.
Вторым по частоте встречаемости признаком является поражение костной системы в виде остеопойкилии (синонимы — врожденная рассеянная склерозирующая остеопатия, врожденная пятнистая множественная остеопатия), возникающей в результате очагового отложения кальция [3, 5]. Признаки остеопойкилии обнаруживаются преимущественно в костях конечностей и плечевого пояса. Клинически они не вызывают жалоб, но важны для дифференциальной диагностики. При рентгенологическом исследовании в костях выявляются мелкие округлые или овальные уплотнения спонгиозных структур костной ткани (рис. 2).
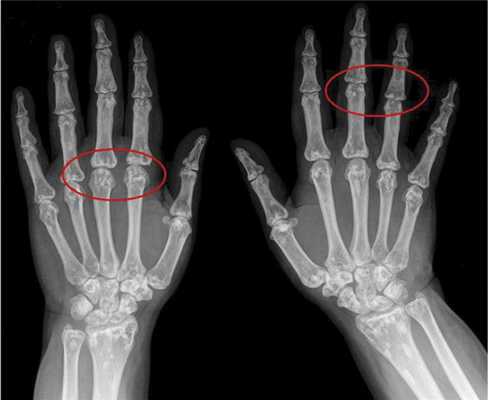
Рис. 2. Остеопойкилия.
Более редкими проявлениями заболевания являются различные пороки развития сердца, ребер, зубов, лицевого скелета, глаз и т. д., эндокринные расстройства (сахарный диабет), неврологические нарушения (эпилепсия, умственная отсталость).
Диагностика синдрома опирается на сочетание типичных клинических симптомов и требует обязательного инструментального подтверждения — морфологического, рентгенологического, генетического. При гистологическом исследовании выявляют признаки соединительнотканного невуса: локальную пролиферацию эластических структур и накопление муцина в дерме. Для подтверждения синдрома Бушке—Оллендорф необходимы дополнительные методы окраски гистопрепаратов по Вейгерту, по Ван-Гизону, толуидиновым синим и сравнение с биоптатом здоровой кожи (рис. 3).
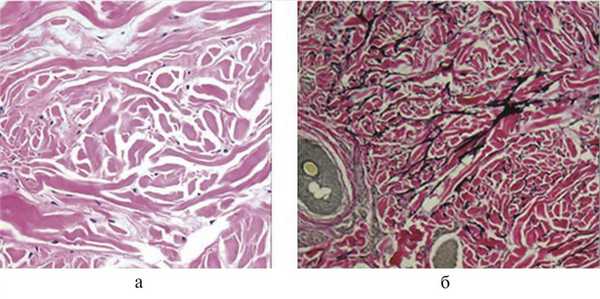
Рис. 3. Морфологическое исследование биоптата (кожа передней брюшной стенки) пациента с синдромом Бушке—Оллендорф. а — участки уплотнения коллагеновых пучков без признаков склерозирования (окраска гематоксилином и эозином; ×40); б — утолщенные пучки эластических волокон, ориентированные в различных направлениях (окраска по Ван-Гизону, ×40).
При невозможности постановки диагноза с помощью рутинных методов применяют молекулярно-генетическую диагностику, заключающуюся в поиске мутаций в гене LEMD3. Течение заболевания в большинстве случаев доброкачественное, хотя возможны и неблагоприятные варианты, приводящие к развитию злокачественных новообразований в области остеопойкилии.
Демонстрацией нашего клинического случая мы хотим показать сложность диагностики и дифференциальной диагностики генодерматозов.
Клинический случай
Пациент А., 5 лет. Ребенок от 6-й беременности (1-я — неразвивающаяся, 2-я и 5-я — доношенный мальчик, 3-я - медицинский аборт, 4-я — выкидыш), протекавшей на фоне угрозы прерывания, анемии, ОРВИ. Роды (третьи) путем экстренного кесарева сечения на 36-й неделе (несостоятельность рубца на матке). Масса тела при рождении 2630 г, рост 48 см, массо-ростовой показатель 55 (норма 60—80), оценка по шкале Апгар 7—8 баллов. Раннее развитие без особенностей. Грудное вскармливание до 18 мес. Вакцинация в соответствии с Национальным календарем прививок. Генеалогический анамнез отягощен по линии матери — аллергические заболевания (атопический дерматит, поллиноз), феномен Вольфа—Паркинсона—Уайта (WPW), артропатии. У старшего сибса пауциартикулярный юношеский артрит, кольцевидная гранулема; у младшего — поллиноз, пищевая аллергия.
Мама считает ребенка больным с рождения, когда после выписки из родильного дома на передней поверхности правого бедра было обнаружено розовое пятно размером до 1 см, округлой формы с четкими контурами. Педиатр на первом патронаже расценил высыпания как проявления аллергической реакции на пищу, рекомендовал маме придерживаться диеты. В возрасте 2—3 мес у ребенка появились эритемато-сквамозные высыпания в области лица (на щеке), туловища, голеней, возникающие на фоне погрешности в питании матери. Элемент на бедре по-прежнему расценивался как проявление пищевой аллергии. Впервые по поводу высыпаний обратились к аллергологу в возрасте 6 мес, был выставлен диагноз атопического дерматита и рекомендована гипоаллергенная диета.
Прогрессирование высыпаний началось после 1,5 лет, когда после перенесенной ОРВИ начали появляться новые элементы, изменился их внешний вид (приобрели желтоватый оттенок) и консистенция (стали возвышаться над поверхностью кожи и уплотнились). Аллерголог по месту жительства назначил терапию топическими глюкокортикостероидами (адвантан) и антигистаминным препаратом 1-го поколения диметинденом (фенистил). Эффекта от терапии не отмечалось. Резкое увеличение количества высыпных элементов отметили в возрасте 3 лет после перенесенной катаральной ангины, а затем через 5 мес после кишечной инфекции (рис. 4).
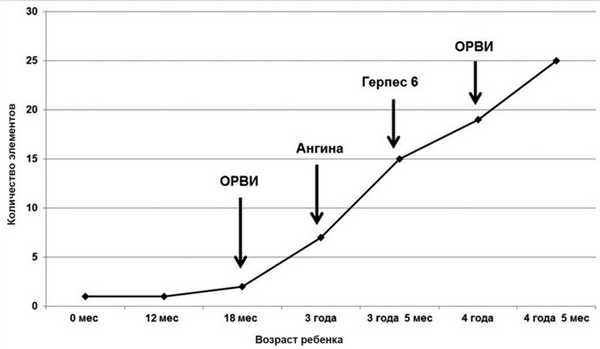
Рис. 4. Динамика кожного процесса. С этого же времени ребенок впервые стал жаловаться на боли в ногах.
За полуторагодовалый период ребенок неоднократно консультирован различными специалистами, проходил сложные и дорогостоящие исследования (табл. 1, 2).
Таблица 2. Проведенные исследования Таблица 1. Консультации специалистов
При осмотре в отделении состояние ребенка удовлетворительное, самочувствие не нарушено. Патологический кожный процесс невоспалительного характера, ограниченный, асимметричный; локализован по передней, боковой и задней поверхностям правого бедра. Представлен подкожными узлами от 0,5 до 1,5 см, желтовато-розового цвета с четкими контурами, полушаровидной формы, с гладкой поверхностью, плотноэластической консистенции, склонными к слиянию (рис. 6).
Рис. 6. Кожный процесс у ребенка А., 5 лет. Задняя и боковая поверхности правого бедра. Пальпация элементов безболезненная; субъективно — зуд после контакта с источником тепла. Кожа вне очагов не изменена, умеренно выражен ксероз. Придатки кожи не изменены, слизистые оболочки интактны. Рис. 5. Рентгенологическое исследование у больного А., 5 лет. а — кисти; б — стопы; в — коленные и голеностопные суставы.
При рутинном обследовании, включавшем клинический анализ крови, общий анализ мочи, биохимическое исследование крови, иммунный статус, патологии не выявлено. При УЗ-исследовании брюшной полости и забрюшинного пространства выявлена ротация обеих почек. Невропатолог диагностировал синдром гиперактивности, аллерголог — пищевую аллергию. На момент осмотра данных, свидетельствующих о синдроме Бушке—Оллендорф, не получено. Заключительный дерматологический диагноз: соединительнотканный невус.
Рекомендации заключались в динамическом наблюдении, фотодокументировании и проведении УЗ-исследования эффлоресценций в области правого бедра 1 раз в 6 мес. По достижении ребенком совершеннолетия возможна хирургическая коррекция, заключающаяся в иссечении невоидных образований. Динамическое наблюдение необходимо, так как возможно отсроченное появление других клинических признаков, входящих в состав синдрома Бушке—Оллендорф, прогноз для выздоровления при этом неблагоприятный. Основной причиной смерти данных пациентов являются онкологические процессы (остеосаркома, хондросаркома), пороки сердца и осложненный сахарный диабет. Своевременная диагностика, грамотная дифференциальная диагностика и правильное ведение таких пациентов необходимо для профилактики осложнений.
Несмотря на схожесть клинических проявлений у ребенка с синдромом Бушке—Оллендорф (типичные кожные проявления), отсутствие типичных поломок в гене LEMD3 позволяет полностью исключить синдром Бушке—Оллендорф. Для определения прогноза необходимо продолжить диагностический поиск (иммуногистохимическое исследование соединительнотканного невуса, полноэкзомное секвенирование фибробластов кожи), что позволит дифференцировать относительно доброкачественные заболевания — множественные дермальные невусы коллагенового или эластинового типов, тяжелого мелореостоза, инвалидизирующего с возрастом.
Синдром Фишера-Эванса
Синдром Фишера-Эванса — это редкое заболевание, представленное комбинацией гемолитической анемии и тромбоцитопении. Патология имеет идиопатический характер, среди провоцирующих факторов выделяют вирусные инфекции, аутоиммунные болезни. Синдром проявляется спонтанными кровотечениями, кровоизлияниями в кожу и слизистые, одышкой при физической нагрузке. Для диагностики назначают клинические, иммунологические и биохимические анализы крови, костномозговую пункцию, генетическое тестирование. Лечение включает глюкокортикоиды, цитостатики, моноклональные антитела, в тяжелых случаях показаны спленэктомия, трансплантация гемопоэтических стволовых клеток (ТГСК).
МКБ-10
Общие сведения
Синдром Фишера-Эванса (СФЭ) — редкая патология, которая встречается с частотой 4,5-20 случаев на 100 тыс. населения, дети болеют чаще взрослых, женщины — чаще мужчин. Пик диагностики приходится на 5-летний возраст, а медиана заболеваемости составляет 58,5 лет по данным датских исследований. До 82% случаев синдрома ассоциированы с различными аутоиммунными нарушениями. Болезнь была впервые описана в 1947 г. Фишером, который предположил ее аутоиммунную природу. Эванс с коллегами в 1951 г. изучили клинику заболевания, подтвердили иммунные механизмы его возникновения.
Причины
При идиопатической форме СФЭ выявить связь гемолиза, тромбоцитопении с другими патологическими процессами в организме не удается. Для симптоматической формы синдрома Фишера-Эванса характерно сочетание с первичными и вторичными иммунодефицитами, перенесенными вирусными или бактериальными инфекциями. Провоцирующим фактором выступают аутоиммунные патологии (СКВ, антифосфолипидный синдром, ревматоидный артрит), лимфопролиферативные процессы.
Патогенез
СФЭ встречается при нарушении иммунной регуляции, снижении толерантности к собственным антигенам тела. Основную роль в патогенезе синдрома играют плазматические клетки, которые вырабатывают антитромбоцитарные антитела, направленные против мембранных гликопротеидов. При этом тромбоциты начинают усиленно разрушаться в селезенке. Деструкция эритроцитов происходит под влиянием аутореактивных IgG (внесосудистый гемолиз), IgA и IgM (внутрисосудистый гемолиз).
В механизме развития синдрома Фишера-Эванса имеет значение нарушение баланса между Т-супрессорами и Т-хелперами с преобладанием последних, что активизирует патологические иммунные реакции. Для СФЭ также характерно возрастание уровней интерлейкина-10 и гамма-интерферона, которые стимулируют В-лимфоциты, усиливают выработку иммуноглобулинов. Важным звеном патогенеза является гиперэкспрессия антигена APO-1, индицирующего апоптоз клеток крови.
Симптомы
Более половины больных страдают одновременно возникающими признаками тромбоцитопении и анемии, в 30% случаев сначала появляется недостаток тромбоцитов, а в 16-25% случаев первыми начинают разрушаться эритроциты. При последовательном развитии цитопений интервал между ними составляет в среднем 4,2 года, но может растягиваться до 16 лет. СФЭ имеет хроническое прогрессирующее течение. Типична манифестация после вирусной инфекции.
Тромбоцитопения характеризуется точечными кровоизлияниями (петехиями), крупными кровоподтеками (экхимозами). Также типичны спонтанные носовые, желудочно-кишечные, маточные, легочные кровотечения. Анемия клинически проявляется бледностью или желтушностью кожи, признаками одышки во время физической нагрузки. К редким признакам болезни относят увеличение лимфоузлов, дискомфорт в животе из-за увеличения печени, селезенки.
Осложнения
Синдром Фишера-Эванса в 55% случаев ассоциирован с нейтропениями и резким снижением иммунитета, что приводит к рецидивирующим инфекциям дыхательной и мочевыделительной системы, тяжелому течению бактериальных процессов, склонности к генерализации воспаления. В 6-14% пациенты с СФЭ страдают от панцитопении — дефицита всех кровяных клеток.
Аутоиммунная анемия при синдроме Фишера-Эванса, как правило, осложняется гемолитическими кризами, а развитие внутрисосудистого гемолиза чревато венозными тромбозами с риском эмболий. При хроническом течении заболевания наблюдается вторичный гемосидероз. Тяжелая тромбоцитопения опасна спонтанными кровотечениями, которые без экстренной медицинской заканчиваются смертью пациента от кровопотери.
Диагностика
Обследование больного с типичными симптомами анемии, тромбоцитопении проводит гематолог. В ходе осмотра врач выявляет внешние признаки дефицита клеточных элементов крови, пальпирует живот, изучает состояние наружных лимфоузлов. Поскольку болезнь Фишера-Эванса считается диагнозом-исключением, специалисту требуется полная информация о состоянии системы крови и всего организма, поэтому он назначает следующие исследования:
- Гемограмма. В результатах анализа определяют умеренную или глубокую тромбоцитопению, нормохромную анемию. На гемолитический характер разрушения эритроцитов указывает повышенный уровень молодых клеток (ретикулоцитов). При длительном течении синдрома в гемограмме обычно обнаруживается нейтропения.
- Проба Кумбса. Исследование считается наиболее информативным для подтверждения иммунного происхождения гемолиза. У 90% больных тест показывает положительный результат. Однако возможны ложноотрицательные показатели, которые вызванные чрезмерной фиксацией антител на эритроцитах.
- Иммунологические исследования. Анализы проводятся для оценки количества сывороточных иммуноглобулинов и их субклассов, соотношения разных типов лимфоцитов. Обязательно исследуют кровь на аутоантитела (антинуклеарные, анти-ДНК, антифосфолипидные), чтобы исключить другие аутоиммунные заболевания.
- Генетические анализы. Молекулярно-генетические тесты необходимы для дифференциальной диагностики болезни Фишера-Эванса с лимфопролиферативным синдромом. В 76% при СФЭ обнаруживают мутации генов FAS, CASP8, CASP10. По показаниям выполняется мультигенная панель иммунодефицитных состояний.
- Костномозговая пункция. Цитология биоптатов костного мозга необходима для исключения злокачественных новообразований системы крови, которые зачастую манифестируют цитопениями. У страдающих синдромом Фишера-Эванса обнаруживают нормальную или повышенную клеточность без признаков атипичного роста.
Инструментальная диагностика не используется для верификации диагноза, но она информативна для установления первопричины синдрома при симптоматическом варианте СФЭ. Для исследования лимфоидных тканей врач выписывает направление на УЗИ лимфатических узлов и селезенки, рентгенографию или КТ грудной клетки. Для уточнения диагноза гематолог направляет пациента на консультацию иммунолога, ревматолога, онколога.
Лечение синдрома Фишера-Эванса
Консервативная терапия
Этиотропное лечение синдрома не разработано, поэтому усилия врачей направлены на коррекцию патологических изменений гемограммы. Назначаемые медикаменты делятся на препараты первой и второй линии. Терапия первой линии рекомендована для быстрой коррекции цитопении, предупреждения жизнеугрожающих осложнений, а лекарства 2 линии используются для поддержания ремиссии.
Схема лечения синдрома Фишера-Эванса начинается с введения глюкокортикоидов в стандартных дозах или в режиме пульс-терапии для быстрого купирования аутоиммунных процессов. При недостаточном эффекте ГКС схему дополняют внутривенными иммуноглобулинами. Чтобы откорректировать последствия массивных кровотечений, назначают трансфузии эритроцитарной взвеси, тромбоцитарного концентрата.
В рамках второй линии терапии активно применяются препараты моноклональных антител (ритусуксимаб, алемтузумаб), которые регулируют аутоиммунные механизмы, уменьшают разрушение компонентов крови. При рефрактерных случаях синдрома показаны цитостатики, обладающие максимальным иммуносупрессивным эффектом. Для удаления части сывороточных антитромбоцитарных иммуноглобулинов эффективен плазмаферез.
Хирургическое лечение
Пациентам, которые не отвечают на медикаментозную терапию, рекомендована спленэктомия. Удаление селезенки повышает шансы достичь длительной ремиссии, а также снижает частоту рецидивов, позволяет уменьшить дозировки глюкокортикоидов. При рефрактерном и беспрерывно рецидивирующем течении синдрома гематологи проводят ТГСК.
Экспериментальное лечение
В последние годы предлагается терапия синдрома Фишера-Эванса препаратами из группы агонистов тромбопоэтиновых рецепторов (элтромбопаг, ромиплостим). Лекарства купируют тяжелую тромбоцитопению, уменьшают риск спонтанных кровоизлияний. Они особенно эффективны в комбинации с кортикостероидами. Для коррекции нейтропении предлагается использовать колониестимулирующие факторы.
Прогноз и профилактика
Синдром Фишера-Эванса отличается хроническим рецидивирующим течением, однако при правильном подборе препаратов удается достичь ремиссии. Прогноз для больного зависит от тяжести цитопении и своевременности начала лечения. Опасения вызывают клинические случаи, когда медикаменты 1-й и 2-й линии не дают позитивного гематологического ответа. Учитывая неясную этиологию синдрома, профилактические меры не разработаны.
1. Синдром Фишера-Эванса: клиническое наблюдение в практике терапевта/ А.А. Якушев, И.Г. Федоров, Л.Ю. Ильченко, С.С. Шмыкова и др.// Архив внутренней медицины. — 2020.
2. Синдром Фишера-Эванса. Клинические рекомендации Национального общества детских гематологов и онкологов. — 2016.
3. Синдром Фишера-Эванса: клинический случай/ Н.Н. Андреева, Н.И. Пенкина, Д.Н. Королева, Г.В. Поверин// Клиническая медицина. — 2019.
4. Синдром Фишера-Эванса у детей: анализ генных нарушений и ответа на терапию/ Ж.А. Кузьминова, Е.Д. Пашанов, А.В. Павлова, М.А. Курникова и др.// Вопросы гематологии/онкологии и иммунопатологии. — 2019.
Кератодермия Бушке-Фишера-Брауэра. Клинический случай
Оленич И.В., Воронцова И.В., Денисова Е.В., Корсунская И.М. Кератодермия Бушке-Фишера-Брауэра. Клинический случай. Дерматология (Прил. к журн. Consilium Medicum). 2017; 3: 46-47.
Olenich I.V., Vorontsova I.V., Denisova E.V., Korsunskaya I.M. Buschke-Fischer-Brauer disease. Clinical case. Dermatology (Suppl. Consilium Medicum). 2017; 3: 46-47.
Ладонно-подошвенная кератодермия Бушке-Фишера-Брауэра - редкое аутосомно-доминантное наследственное кожное заболевание, характеризующееся множественными гиперкератотическими папулами на ладонях и подошвах. Мутация генов локализуется во втором локусе 15q22.2-15q22.31 хромосом. В данной работе приводятся описание клинического случая пациента и рекомендованная терапия.
Buschke-Fischer-Brauer disease is a rare autosomal dominant disorder of keratinization, characterized by multiple hyperkeratotic lesions on the palms and soles. Mutation of genes localized in the second locus 15q22.2-15q22.31 chromosomes. This paper provides a description of a clinical case of the patient and the recommended treatment.
1. Oztas P, Alli N, Polat M et al. Punctate Palmoplantar Keratoderma (Brauer-Buschke-Fischer Syndrome). Am J Clin Dermatol 2007; 8: 113-6.
2. Gao M, Yang S, Li M et al. Refined localization of a punctate palmoplantar keratoderma gene to a 5.06-cM region at 15q22.2-15q22.31. Br J Dermatol 2005; 152: 874-8.
3. Itin PH, Fistarol SK. Palmoplantar keratodermas. Clin Dermatol 2005; 23: 15.
Читайте также:
- Общие сведения о смерти и умирании
- Лапароскопические операции у детей. Операция при паховой грыже, гидроцеле и перекруте яичка у детей
- Синдром Воренже (Woringer)
- Оборудование для оценки маточно-плацентарного кровообращения. Методика оценки маточно-планцентарного кровообращения
- Фиксация центральной окклюзии по Сидоренко. Анатомическая постановка искусственных зубов.