Синдром липидоза - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 22.01.2026
Научно-исследовательский институт нефрологии ГБОУ ВПО "Санкт-Петербургский государственный медицинский университет им. акад. И.П. Павлова"
Научно-исследовательский институт нефрологии ФГБУ ВО ПСПбГМУ им. И.П. Павлова Минздрава РФ, Санкт-Петербург, Россия
Волгоградский государственный медицинский университет, Волгоград, Россия
Случай диагностики липопротеиновой гломерулопатии в России
Липопротеиновая гломерулопатия (ЛПГ) — редкое заболевание, характеризующееся особенными гистологическими, иммуноморфологическими и ультраструктурными изменениями. Основными патоморфологическими признаками ЛПГ являются липопротеиновые тромбы в просвете капиллярных петель, протеинурия и дислипопротеинемия в виде повышения концентрации аполипопротеина Е (фенотипа Е2/Е3, Е2/Е4). Пациент 47 лет имел нефротический синдром, суточная потеря белка 12,4 г/сут. Исследование нефробиоптата на светооптическом уровне выявило судан III-позитивные, слабо PAS, эозин-позитивные рыхлые тромбы в просвете капиллярных петель. Иммуноморфологически в тромботических массах отложений иммуноглобулинов не выявлено. Ультраструктура тромботических масс состояла из гранулярных масс с многочисленными вакуолями, формирующих концентрические слои. Таким образом, была диагностирована ЛПГ как проявление гиперлипопротеинемии 3-го типа, которая имела характерные патоморфологические и клинико-лабораторные признаки данного заболевания.
Дислипопротеинемия является независимым предиктором снижения функции почек в общей популяции условно здоровых людей и ассоциируется с многими гемодинамическими, структурными и гистопатологическими альтерациями почек на фоне метаболических сдвигов, которые предшествуют этим повреждениям [1, 2]. Одним из редких вариантов нарушения обмена липидов является липопротеиновая гломерулопатия (ЛПГ). ЛПГ — заболевание с аутосомно-рецессивным наследованием, характеризующееся оригинальными гистопатологическими признаками в виде липопротеиновых тромбов и гиперлипопротеинемией (ГЛП) (3-й тип ГЛП по Фредериксону), с нарушением соотношений фракций липидов, в частности увеличением концентрации сывороточного аполипопротеина Е (АроЕ) — фенотип Е2/4 или Е2/3.
Впервые ЛПГ как отдельная нозологическая форма гломерулярной патологии была представлена японскими исследователями в 1989 г. [3, 4], которые на основании анализа ряда случаев с общими признаками: липопротеиновые тромбы, дислипидемия, соответствующая 3-му типу классификации гиперлипидемий [5], предложили название для данной патологии — ЛПГ. В 1995 г. это заболевание было включено в Классификацию и Атлас гломерулярных болезней — Classification and Atlas Glomerular Disease [6], изданные совместно с ВОЗ. Позже была установлена связь нового заболевания с мутациями гена apoE, что позволило определить наследуемый риск развития данного заболевания.
К настоящему времени во всем мире описано около 100 случаев ЛПГ. Большинство пациентов были из Японии, Китая, Тайваня и США. Несколько случаев было отмечено в Европе, в частности во Франции, Италии [7] и Латинской Америке [8]. Заболеванием страдают преимущественно мужчины среднего возраста, хотя могут болеть женщины и дети, причем болезнь длительное время может протекать без клинических проявлений. После нескольких лет бессимптомного течения отмечается развитие прогрессирующей протеинурии с формированием нефротического синдрома, повышением артериального давления и снижением скорости клубочковой фильтрации.
В связи с реальными сложностями клинической и патоморфологической диагностики из-за крайне редкой встречаемости заболевания мы решили представить пациента, находившегося в клинике кафедры пропедевтики внутренних болезней в 2007 г., в качестве диагностического примера.
Больной Г., русский, 47 лет, поступил в клинику 16.05.2007 для дополнительного обследования из Калининградской больницы. Из анамнеза известно, что в декабре 2006 г. обратился с жалобами на появление отеков лица и нижних конечностей, повышение артериального давления до 190/130 мм рт.ст. В анализах мочи: креатинин крови 134—122 мкмоль/л, холестерин 10 ммоль/л, общий белок 50,7 г/л; общий анализ мочи: протеинурия 0,066 г/л, лейкоцитурия 2—4 в поле зрения (п/зр.), эритроцитурия 0—1 п/зр.
При поступлении в клинику пропедевтики внутренних болезней СПбГМУ им. акад. И.П. Павлова жалобы на отеки лица по утрам и нижних конечностей к вечеру, повышение артериального давления до 190/130 мм рт.ст. без субъективных ощущений, тошноты, рвоты, потери сознания. Объективно: состояние удовлетворительное, сознание ясное, кожные покровы чистые, суставы не изменены.
Подкожно-жировая клетчатка развита удовлетворительно, пастозность голеней. Пульс 78 в минуту, ритмичный, обычных свойств. Границы абсолютной и относительной сердечной тупости не изменены. Тоны сердца чистые. Артериальное давление 180/110 мм рт.ст.
При перкуссии ясный легочный звук, границы легких не изменены. Дыхание везикулярное, хрипов нет. Язык влажный, чистый. Живот мягкий, безболезненный, печень не увеличена. Клинический анализ крови: Hb 116 г/л, эр. 3,9∙10 12 /л, цв. пок. 0,89, тр. 260,0∙10 9 /л, л. 5,7∙10 9 /л, э. 1%, п. 2%, с. 66%, л. 29%, мон. 2%; СОЭ 60 мм/ч. Биохимическое исследование сыворотки крови. Общий белок крови 40 г/л, альбумины 17 г/л, К 4,3 ммоль/л, Nа 141 ммоль/л, Са 2+ 1,77 ммоль/л, креатинин крови 0,164 ммоль/л, протромбиновый индекс 106%, фибриноген 6,66 г/л. Пробы на HCVAb и HbsAg отрицательные, антинуклеарный фактор 1
Результаты исследования нефробиопсии
Светооптически в срезах корковый слой с числом клубочков до 23, один склерозирован полностью. Клубочки значительно увеличены в размерах. В большинстве клубочков умеренная пролиферация клеток мезангия с увеличением мезангиального матрикса (рис. 1, а). В мезангии фуксинофильные депозиты не выявлялись. Просвет капилляров растянут за счет накопления в просвете рыхлых, местами вакуолизированных не фуксинофильных, PAS-слабоположительных, слабо эозинофильных тромбов, имеющих диффузно-глобальную локализацию, практически обтурирующих просвет петель (см. рис. 1, а, б). Кроме того, в просвете капилляров встречались единичные эритроциты и лейкоциты. Базальные мембраны капилляров клубочков местами утолщены и расщеплены (см. рис. 1, в). Зернистая и гиалиново-капельная дистрофия отмечалась в эпителии канальцев. Межтубулярная строма с очагами склероза и лимфогистиоцитарной инфильтрации. Выявлялся умеренный периваскулярный склероз и очаговый заместительный склероз с участками атрофии и субатрофии прилежащих канальцев. В сосудах мышечного типа гипертрофия мышечного слоя. Реакция конго красным отрицательная.
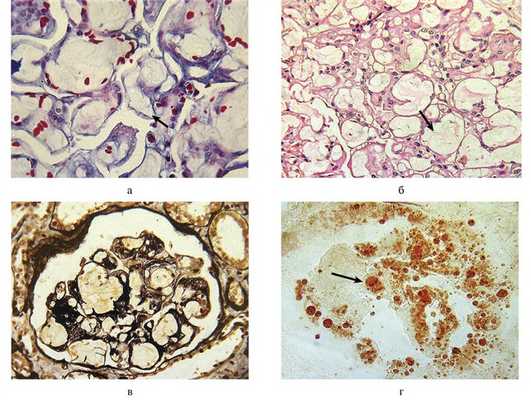
Рис. 1. Светооптическая морфологическая характеристика клубочка почки у больного Г. а — диффузное расширение капиллярных петель клубочка за счет обтурирующих, рыхлых, нефуксинофильных тромбов. Диффузно-глобальная локализация. Единичные эритроциты в просвете капилляров. Окраска по Массону, ×350; б — резкое расширение мезангиального матрикса с умеренной гиперклеточностью. Просвет капилляров клубочков резко расширен, заполнен рыхлыми слоистыми эозинофильными массами (черная стрелка). Окраска гематоксилином и эозином, ×200; в — мембраны клубочка утолщены и местами удвоены (розовая стрелка). В просвете капилляров аргиронегативные рыхлые массы. Окраска по Джонсу, ×180; г — отчетливая локализация липидов в просвете капилляров клубочка. Окраска суданом III (черная стрелка), ×180.
При гистохимической окраске суданом III на криостатных срезах в просвете расширенных капилляров располагались жировые включения, окрашенные в оранжевый цвет (см. рис. 1, г).
При иммунофлюоресцентном исследовании обнаружены мезангиальные и отложения IgM (2+), С3 (2+), а также располагавшиеся вдоль базальных мембран расширенных капилляров клубочков. IgA и IgG (+) располагались сегментарно по базальным мембранам клубочка (рис. 2, а, б).
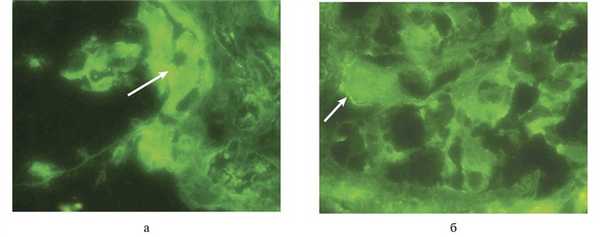
Рис. 2. Иммунофлюоресцентное исследование. а — очаговые линейные отложения IgM (белая стрелка) сегментарно вдоль базальных мембран и частично в мезангии капилляров клубочка. Метод ПИФ, FITC, ×640; б — отложения С3-фракции комплемента (белая стрелка) в виде сегментарных, линейных, субэндотелиальных и частично мезангиальных зонах капилляров клубочка. Метод ПИФ, FITC, ×640.
При ультраструктурном исследовании наблюдались признаки слияния цитоподий, цитоплазматические вакуоли подоцитов и очаговое расщепление базальных мембран (рис. 3, а). В просвете капиллярных петель располагались липопротеиновые тромбы, состоявшие из концентрических слоев вакуолей различного размера, напоминавшие местами «отпечатки пальцев». Просвет капилляров заполнен этими массами с секвестрированием в них эритроцитов. Аналогичный вакуолизированный материал местами отмечался в мезангиальном матриксе (рис. 3, б).
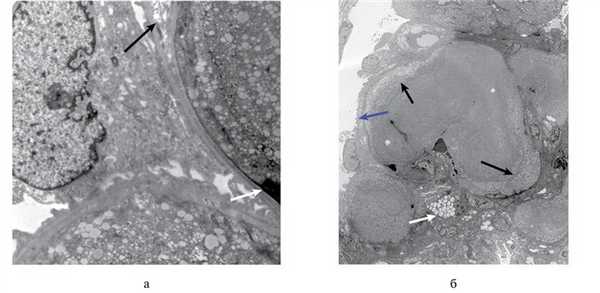
Рис. 3. Трансмиссионная электронограмма. а — вакуоли в просвете капилляра формируют ламинарные слои «липопротеинового тромба». Участок расщепления базальных мембран (черная стрелка). Субэндотелиальный депозит (белая стрелка), ×9500; б — интракапиллярные тромбы, состоящие из концентрических слоев (черные стрелки) мелковакуолизированного материала — «липопротеиновые тромбы». Локально вакуолизированный материал располагается в мезангии (белая стрелка) и в участках расщепления базальных мембран (синяя стрелка), ×3000.
ЛПГ — это новая клиническая нозология, имеющая типичные морфологические изменения, связанные с накоплением в структурах клубочка аполипопротеина Е (АЛП-Е) и В (АЛП-В) при изменении липопротеинового профиля плазмы крови. В частности, последний характеризуется высоким уровнем триглицеридов — холестерина во фракциях липопротеинов очень низкой (ЛПОНП) и промежуточной плотности.
При этом выявляется высокий уровень АЛП Е (двукратное превышение нормы) в сыворотке крови. АЛП-Е состоит из 299 аминокислот с ЛПНП-рецепторсвязующим N-терминалом и липидсвязующим С-терминалом.
Фенотип Апо-Е определяемый изоэлектрическим фокусирующим полиакриламидным гель-электрофорезом, является гетерозиготным для Е2 (Е2/3, Е2/4 и др.), что характерно для ЛПГ в отличие от семейной 3-го типа ГЛП (гомозиготной по Е2). Эти изоформы играют существенную роль, опосредуя взаимодействие липопротеинов с клеточными рецепторами: рецептором ЛПНП и рецептор-связанным протеином ЛПНП, модулируя таким образом катаболизм различных липопротеинов [5]. Однако только у 5% людей, имеющих гомозиготный фенотип Е2/Е2, развивается 3-й тип ГЛП, что свидетельствует о важности других генетических или приобретенных факторов, способствующих развитию данной патологии. Среди фенотипов АЛП-Е при 3-м типе ГЛП Е3/Е3 встречается редко. Для ЛПГ характерна высокая частота встречаемости фенотипа Е2 в виде Е2/3 или Е2/Е4 [9, 10].
У больных выявляется стероидоустойчивый нефротический синдром с суточной потерей белка от 0,3—18 г/сут. Гематурия обычно отсутствует, а системные проявления, такие как гипертензия, атеросклероз, дисфункция печени и другие умеренно выражены. Избирательное вовлечение в процесс преимущественно почек, а не других органов, по-видимому, может быть связано со специфическими взаимодействиями вариантов АЛП-Е с неустановленными на настоящий момент элементами гломерулярной эндотелиальной стенки. Хотя некоторые авторы полагают, что в процессе накопления липидов в клубочках определенную роль могут играть мезангиоциты, которые посредством Fc-рецепторов участвуют в регуляции клиренса ЛПОНП в клубочках [11, 12]. Возраст больных может колебаться от 4 до 69 лет, соотношение мужчин/женщин 3/2. В некоторых случаях ЛПГ может быть ассоциирована с другими гломерулопатиями: IgA-нефропатией, мембранозным гломерулонефритом, системной красной волчанкой [13]. В семейном анамнезе в большинстве случаев патология почек отсутствует [14].
Патогистологическое исследование обеспечивает базовую диагностическую информацию. Типичные признаки выявляются при светооптическом, электронно-микроскопическом и иммуноморфологическом исследованиях, что и наблюдалось в нашем случае. Так, патоморфологические признаки при ЛПГ характеризовались прежде всего изменениями клубочков. В клубочках отмечалось увеличение размера последних за счет диффузно-глобального аневризматического расширения капилляров (см. рис. 1, а). Дилатированные капилляры заполнены бледно окрашенной мелко вакуолизированной субстанцией ламеллярного строения, которая слабо окрашивалась эозином, PAS (см. рис. 1, б). При окраске суданом III интракапиллярные тромбы имели позитивное окрашивание (см. рис. 1, г). В мезангии умеренное сегментарное увеличение матрикса и клеточности, а также мезангиолизис [15]. Стенки капилляров имели сегментарные утолщения и расщепления базальных мембран (см. рис. 1, в). В тубулоинтерстиции исследуемых нефробиоптатов патоморфологические изменения характеризовались очагами склероза с умеренной лимфогистиоцитарной инфильтрацией и соответствующей атрофией и субатрофией прилежащих канальцев, а также с гиалиново-капельной, вакуолярной дистрофией тубулярного эпителия.
Иммуноморфологически наиболее характерным является отложение IgM, C1q по периферии капиллярных петель, хотя не исключается локализация и других иммуноглобулинов и фракции комплемента мезангиальной локализации. В нашем случае выявлены очаговые мелкогранулярные, сегментарные отложения IgM, IgG и С3 в мезангиальном матриксе и вдоль базальных мембран (см. рис. 2, а, б).
Ультраструктурный анализ обычно позволяет уточнить специфические структурные изменения в клубочках при ЛПГ. В частности, в просвете исследованных капилляров у нашего больного наблюдались тромбоподобные субстанции, состоящие из характерных для данной патологии вакуолей различного размера, формировавших концентрические слои, иногда напоминающие «отпечатки пальцев» (см. рис. 3, б). В периферических базальных мембранах отмечалось их сегментарное расщепление (см. рис. 3, а). Липидный материал накапливался не только в просвете капилляров, но также субэндотелиально и в мезангиальном матриксе (см. рис. 3, б). Для подоцитов характерно слияние отростков и накопление в цитоплазме умеренного количества липидных вакуолей. Кроме того, очаговые электронно-плотные отложения наблюдались в мезангии и субэндотелиально.
Таким образом, на настоящий момент основными критериями диагноза ЛПГ являются данные клинического, патологического и лабораторного обследования больного. К таковым в настоящее время относятся умеренная или тяжелая степень протеинурии, расширенные капилляры с бледно окрашиваемой субстанцией в просвете капилляров петель клубочков, песочного вида гранулы в просвете капиллярных петель (при ультраструктурном исследовании так называемые липопротеиновые тромбы), 3-й тип дисбаланса липопротеинов с увеличением количества АроЕ с фенотипом Е 2/3 или Е 2/4 иногда и с другими формами [9, 10].
АроЕ имеет массу 34 кДа, его ген локализуется в 19q 13.2 хромосоме с четырьмя экзонами и тремя интронами [16]. У людей существует 3 общих аллеля ε 2, ε 3, ε 4, соответственно они синтезируют АЛП 2, 3, 4. Эти три формы различаются полиморфностью сайтов, аргинина или цистеина в кодоне 112 и 158. Причем связующая активность с липопротеиновыми рецепторами у АЛП 2 составляет 1% от таковой у АЛП 3 или АЛП 4. На сегодняшний день установлен полиморфизм 54 одиночных нуклеотидов у человека. Полиморфные формы АЛП-Е играют важную роль в патогенезе прогрессии различных почечных заболеваний. Они могут регулировать функции мезангия, повышать риск сердечно-сосудистых заболеваний у уремических пациентов, могут предрасполагать к развитию диабетической нефропатии, а также влиять на тяжесть течения гломерулопатий и ответ на терапию [16]. Мутации структуры АЛП-Е впервые установлены в 1997 г. посредством ДНК-анализа, выявившего замену нуклеотидов G на С в кодоне 145 АЛП-Е. Эта мутация означала замену пролина на аргинин в позиции 145 АЛП-Е. Вариант АроЕ (Arg145 Pro) был назван аро Sendai в честь города, где проживал пациент с ЛПГ. Впоследствии выявлены другие типы мутаций: АроЕ: Kyoto (Cys 25 Arg), ApoE Tokyo (Leu 141 Lys143 del), ApoE (Gln 156 — Gly 173 del), ApoE Maebashi (Arg 142 Leu 144 del), ApoE Guangzhou (Arg 150 pro), ApoE Chicago (Arg 147 Pro). При этом морфологические изменения не зависят от вариантов данных мутаций и остаются стандартными. Более, того у некоторых пациентов уровень АЛП-Е в сыворотке крови может быть нормальным [17].
Эти открытия мутаций АЛП-Е позволяют предположить, что данные изменения играют каузативную роль в возникновении и развитии ЛПГ. Имеется ввиду то обстоятельство, что, например, мутации АЛП-Е АроЕ (Arg145 Pro) в области рецепторсвязующего домена могут приводить к уменьшению рецепторсвязующей активности и предрасполагать к патологическому накоплению АЛП-Е-агрегации и осаждению последних на капиллярной стенке или в мезангии [18].
С другой стороны, вариант мутации Kyoto (Cys 25 Arg) располагается далеко от рецепторсвязующего домена и сопровождается системным липидозом и гиперлипидемией, демонстрируя повышенную связующую активность эндотелия капилляров клубочка [17]. В последних исследованиях [19] установлено, что комбинация аллелей, например АpoE Tokyo/Maebashi и apoE2 (Arg158Cys), вносит дополнительные изменения в патоморфологическую картину заболевания.
Заключение
Таким образом, мы диагностировали с использованием различных патоморфологических методик и клинико-лабораторных данных у больного Г. ЛПГ как проявление ГЛП 3-го типа, которая имела все патогномоничные признаки, изложенные в данной демонстрации. Ретроспективно можно отметить, что у больного после полученной патогенетической терапии отмечена положительная динамика клинико-лабораторных показателей, и он был выписан из стационара. Однако в дальнейшем, на наш взгляд, необходимо проведение дополнительного молекулярно-генетического обследования на предмет идентификации генотипа мутации АроЕ и длительное наблюдение и обследование данного пациента.
Сфинголипидозы
Сфинголипидозы - это группа липидозов, при которой имеет место нарушение обмена сфингомиелина с накоплением его в мозге и внутренних органов, прежде всего - в системе мононуклеарных фагоцитов. В стернальном пунктате выявляются специфические клетки Нимана-Пика.
Болезнь Фабри - форма липидоза, характеризующаяся накоплением гликолипида церамидтригексозида в стенках кровеносных сосудов, в периферических нервах, в нейронах мозга, в роговице, в почках. Развивается вследствие недостаточности фермента α-галактозидазы (А). Заболевание полисистемное. Проявляется наличием ангиом, кератом, поражением роговиц, сенсорной и вегетативной ганглиополиневропатией с пароксизмальными парестезиями и болями в конечностях. Отложение гликолипидов в сосудах может вызвать нарушение кровообращения и, в частности, развитие мозговых инсультов, почечную недостаточность, поражение других органов.
Крабе лейкодистрофия - проявляется в возрасте 2-6 месяцев. Сначала наблюдается остановка, а затем редукция физического и психического развития. При этом характерны приступы крика, центральный парез мышц конечностей, гиперестезия, позже - приступы мышечных спазмов, опистотонус с вытянутыми и перекрещенными ногами, клонические судороги; атрофия зрительных нервов, сопровождающаяся прогрессирующим снижением зрения; гипертермия. Прогноз плохой. В типичных случаях болезнь длится 5-8 месяцев. В терминальной стадии отмечается слепота, кахексия, судороги, децеребрационная ригидность, слабоумие.
Болезнь Ниманна-Пика - группа липидозов, при которых имеет место нарушение обмена сфингомиелина с накоплением его в мозге и внутренних органах. Классическая форма болезни проявляется в возрасте 5-6 месяцев расстройствами зрения и слуха, умеренных повышений мышечного тонуса, вскоре сменяющихся мышечной гипотонией, а затем атонией. Олигофрения, чаще в форме идиотии. Характерны желто-коричневый цвет кожи, гепатоспленомегалия, увеличение лимфатических узлов.
Лейкодистрофии (дегенеративное заболевание мозга с преимущественным поражением белого вещества) - это наследственные диффузные демиелинизирующие процессы в белом веществе головного мозга, спинного мозга и периферических нервов, имеющие прогрессирующее течение. В основе их - генетически детерминированное нарушение обменных процессов в миелиновых структурах и распад неправильно сформированного миелина (дисмиелинизация).
Метахроматическая лейкодистрофия (сульфатиоз, Шольца синдром) - чаще других встречающаяся форма лейкодистрофии, обусловленная редукцией активности лизосомного фермента арилсульфатазы (А) (цереброздсульфатаза) в связи с дефектом фермента на 22-й хромосоме. При этом происходят метахроматически окрашивающиеся вещества - в мозге, печени, почках, уменьшение в мозге количества цереброзидов. В результате происходит диффузная демиелинизация нервной ткани, накопление гранул сульфатидов в клетках глии, макрофагах, в нейронах, поражение ганглиозных клеток сетчатки, избыток липидов в печени, в почечных канальцах.
Заболевание проявляется в возрасте от 1 года до 30 лет (1-4 года - инфантильная форма, 5-20 лет - ювенильная форма, позже 20 лет - взрослая форма). Нарастает мышечный тонус, нарушается координация движений, изменяется походка. Появляются глазодвигательные расстройства, дизартрия, снижаются сухожильные рефлексы. Грубо нарушается психическое развитие, могут быть судорожные припадки. В далеко зашедшей стадии (через несколько лет) развиваются тетраплегия, децеребрационная ригидность, декортикация, грубые псевдобульбарные и бульбарные нарушения. На КТ головного мозга обнаруживают симметричные очаги пониженной плотности в белом веществе лобных и теменных долей мозга. В мозге выявляется почти полная демиелинизация с дегенерацией демиелинизированных волокон, особенно выраженная в каудальной части мозгового ствола. Смерть наступает через 2-10 лет после начала заболевания.
Лейкодистрофия Ван-Богарта - Шерара-Эпстайна - характеризуется нарастающим слабоумием, эпилептиформными приступами, экстрапирамидными симптомами, псевдобульбарными расстройствами. Церебральный холестериноз, сопровождающийся отложением холестерина в двигательной зоне коры большого мозга, ножках мозга и мозжечка, а также отеком клеток передних рогов спинного мозга. Проявляется задержкой физического и психического развития, сухостью кожи, слабостью лицевой мускулатуры, диазатрией, мозжечковой атаксией, бульбарными и пирамидными расстройствами, катарактой, остеопорозом, ксантомой.
Болезнь Вольмана - ранняя инфантильная органомегалия, рвота, диарея, различные повреждения нервной системы, желтуха. Дефектный фермент - кислотная липаза, накапливаемое вещество - эфир, холестерола, триглицириды.
Липоидозы ( Лизосомные болезни накопления липидов , Липидозы , Ретикулоэндотелиозы )
Липоидозы - наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов и их метаболитов в различных органах и тканях. Общие клинические проявления представлены прогрессирующим расстройством интеллектуальных и двигательных функций, поражением костей, кожи, центральной нервной системы, глаз и внутренних органов (печени, почек, селезенки). Диагностика основана на лабораторных исследованиях ферментативной активности, количества токсичного субстрата, наличия мутации в генах. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
МКБ-10
Общие сведения
Синонимичные названия липоидозов - липидозы, ретикулоэндотелиозы, лизосомные болезни накопления липидов. К данной группе относится сфингомиелиноз, болезнь Тея-Сакса, болезнь Гоше, семейные гиперлипидемии и некоторые другие заболевания. Общей характеристикой является патологическое накопление липидов внутри клеток организма в результате дефекта ферментных систем. Липоидозы относятся к группе редких (орфанных) заболеваний. Их распространенность очень низка, для отдельных типов патологий составляет от 1:40 тыс. до 1:1 млн. и реже. Суммарная частота - 1:7 тыс. Большинство липидозов имеют прогредиентное течение, приводят к инвалидизации и ранней смерти.
Причины липоидозов
Определяющим фактором развития липидозов является генетический дефект, который обуславливает полную или частичную недостаточность лизосомальных ферментов, расщепляющих сложные липиды. Наследование болезней происходит по аутосомно-рецессивному механизму. Это означает, что новорожденный оказывается больным, если получает мутационный ген от каждого родителя (имеет пару дефектных генов в аллели). Когда мутация передается только от матери или отца, то ребенок является ее носителем и остается здоровым.
Исключительный механизм передачи дефекта у болезни Фабри. В отличие от других липидозов, она наследуется по X-сцепленному рецессивному типу. Гемизиготные пациенты мужского пола больны, передают мутацию только дочерям. У девочек заболевание всегда проявляется при наличии двух рецессивных (измененных) генов. Иногда симптомы липидоза проявляются у пациенток с одним дефектным геном, когда доминантный ген оказывается инактивированным (причины этого не выяснены).
Патогенез
Патогенетической основой большинства липоидозов является генетически детерминированный ферментативный дефект - энзимопатия. Вследствие этого лизосомы клеток накапливают липиды и промежуточные продукты их метаболизма, что ведет к прогрессивному нарастанию нарушения функций органов. При синдроме Вольмана определяется недостаточность кислой этеразы, становится невозможным полный цикл обмена холестерола, повышается содержание его эфиров в лизосомах селезенки, печени, надпочечников, кишечника и костного мозга. У пациентов с болезнью Гоше снижено или полностью отсутствует производство бета-глюкозидазы; в печени, селезенке, костном мозге скапливаются продукты расщепления сфинголипидов. Болезнь Нимана-Пика развивается на базе дефицита сфингомиелазы, характеризуется повышением количества сфингомиелина во многих клетках, особенно в гепатоцитах, нейронах. При идиотии Тэя-Сакса существует дефект N-ацетилгексозаминидазы, происходит накапливание ганглиозидов в головном мозге.
Классификация
Наследственные нарушения метаболизма сложных липидов представлены группой различных по происхождению заболеваний, которые на патогенетическом уровне связаны с патологией жирового обмена. Для липоидозов характерно скопление сложных липидных соединений внутри лизосом клеток. В зависимости от того, какой липид не расщепляется до конца и откладывается в тканях, выделяют несколько типов болезней:
- Гликолипидозы. При данном типе заболеваний невозможен полный распад гликолипидов - соединений, состоящих из углеводов и жирных кислот. Гликолипидозы представлены цереброзидозами (сфинголипидоз Гоше, болезнь Краббе), сульфатидозами (метахромотическая лейкодистрофия), церамидолигозидозами (болезнь Фабри, церамидлактозидлипоидоз), ганглиозидозами (болезнь Сандхоффа, ранняя детская амавротическая идиотия).
- Липопротеинемии. Обусловлены патологией обмена липидов плазмы крови, основанной на генетическом дефекте ферментов или рецепторов клеток. Липиды плазмы - жирные кислоты, триглицериды, холестерин. Липопротеинемии включают семейную гиперхолестеринемию, комбинированную семейную гиперлипидемию и семейные гиперлипидемии типов I-V.
- Сфингомиелиноз. Синонимичное название - болезнь Ниманна-Пика. При развитии этого заболевания в ретикулоэндотелиальных клетках увеличивается содержание фосфолипидного соединения сфингомиелина.
Симптомы липоидозов
Клиническая картина липидозов определяется особенностями вовлечения в патологический процесс органов и систем. При болезни Гоше I типа поражается печень, селезенка, кости и костный мозг. Симптомы включают увеличение размеров печени, хрупкость костей, анемию, лейкопению, снижение свертываемости крови. II тип заболевания разворачивается с преимущественным повреждением ЦНС и печени. Наблюдаются судорожные приступы, мышечный гипертонус, спастичность, интеллектуальная недостаточность, расстройства акта глотания. У людей с галактозилцерамидным липидозом (болезнью Краббе) снижается функциональность миелиновой оболочки. Развивается гипервозбудимость, рвота, судороги, задержка психомоторного развития с прогрессирующим снижением интеллекта и зрения.
При метахромотической лейкодистрофии патологически изменяется миелиновая оболочка. Основные симптомы - гипотония мышц рук и ног, снижение (отсутствие) сухожильных рефлексов, атаксия, атрофия зрительных нервов, нистагм, спастический тетрапарез, глухота, интеллектуальное и моторное недоразвитие. Клинические признаки болезни Андерсона-Фабри разнообразны, одними из наиболее распространенных являются нейропатическая боль (умеренной и легкой выраженности, в ступнях и ладонях) и кризы Фабри - сильные жгучие боли в конечностях приступообразного характера. Дополнительно возникает гипогидроз или ангидроз, непереносимость физических нагрузок, ангиокератомы, расстройства слуха, сердечно-сосудистой и почечной функций.
При развитии синдрома Сандхоффа определяется общая вялость, гипотонус мышц конечностей, трудности сосания и глотания, прогрессирующая задержка моторного и психического развития, двигательная слабость, шумы в сердце, судороги, слепота, увеличение селезенки. При ранней детской амавротической идиотии больше всего поражается центральная нервная система. К концу первого полугодия ухудшается реакция детей на внешние сигналы (лица близких, игрушки), утрачиваются двигательные навыки, снижается познавательный и игровой интерес. Нарушается зрение вплоть до слепоты. Формируются судорожные припадки.
Первые симптомы сфингомиелиноза - вялость, малоподвижность, апатичность, отказ от еды, рвота. Позже увеличивается живот (гепатомегалия), конечности становятся худыми, кожа приобретает коричневатый оттенок, периоды заторможенности сменяются гипервозбуждением. Дети отстают в психофизическом развитии. Диагностируется умеренная гидроцефалия, гипертермия, спастический парез ног и рук, приступообразная асфиксия.
Проявления гиперлипопротеинемий обнаруживаются в возрасте до 10 лет. Для всех форм болезней свойственно отложение подкожного жира в виде ксантом, а также абдоминальные боли, панкреатит, гепатоспленомегалия (скопление липидов в селезенке, печени). При комбинированной семейной гиперлипидемии и гиперлипидемии II типа возможен ранний атеросклероз сосудов. При гиперлипидемиях IV и V типа - снижение чувствительности к глюкозе, ИБС.
Осложнения
Липоидозы протекают с развитием недостаточности жизненно важных органов. Наиболее частыми осложнениями становятся заболевания сердца и сосудов, ЦНС, почек, легких, печени. Пациенты страдают от атеросклероза сосудов, сердечной и дыхательной недостаточности, ишемической болезни сердца, хронической почечной и печеночной недостаточности, аденом и цирроза печени, инсультов, транзиторных ишемических атак. При типах заболеваний, совместимых с жизнью, зачастую нарушено двигательное и психическое развитие. Многие больные оказываются неспособны к самообслуживанию, обучению и овладению профессией, нуждаются в пожизненном уходе со стороны окружающих.
Диагностика
Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением занимаются педиатры, гематологи, гастроэнтерологи, ревматологи, неврологи, психиатры и врачи-генетики. На первичном этапе диагностики проводится сбор семейного анамнеза: при наследственном характере ферментопатии у больного могут быть родственники с подтвержденным диагнозом липидоза. При сборе клинических данных специалисты обращают внимание на время начала симптомов: чаще всего болезнь проявляется в период новорожденности или первого года жизни, редко - у детей постарше или взрослых. К специфическим методам обследования пациентов относят:
- Анализ активности дефектного фермента. Исследованию подвергаются различные биоматериалы - плазма крови, лейкоциты, сухие пятна крови, культура фибробластов кожи, биопсийный материал почек, печени. При липоидозах определяется недостаток активности определенного фермента: от легкого снижения до полного отсутствия.
- Количественное исследование липидов. В крови и биопсийном материале органов анализируется содержание патологически накапливаемых липидов и промежуточных продуктов их обмена. У больных липоидозом показатели превышают норму. Параллельно изучаются изменения строения пораженных клеток.
- Секвенирование ДНК. Выявление дефектного гена в хромосоме является наиболее точным, но трудоемким методом диагностики наследственных болезней. Широко применяется в рамках перинатальной и преимплантационной диагностики, в случаях, когда вышеперечисленные анализы не дают однозначного представления о диагнозе.
- Визуализирующие исследования органов. Дополнительно проводится диагностика состояния пораженных органов - сердца, печени, желчного пузыря, селезенки, легких, почек, головного мозга. Используется УЗИ, МРТ, КТ, ЭКГ, ЭЭГ. Процедуры позволяют оценить размеры, выявить структурные изменения и новообразования в органе.
Лечение липоидозов
Терапия данной группы заболеваний - сложная задача для врачей разных специальностей. Методы лечения несовершенны и продолжают разрабатываться, при некоторых видах болезней возможно добиться лишь временного улучшения самочувствия больного, при других достижима стойкая ремиссия. Общая схема медицинской помощи больным состоит из трех компонентов:
- Ферментозаместительная терапия. В организм пациентов вводятся препараты с искусственно выделенным дефицитарным ферментом. Инъекции выполняются пожизненно, позволяют восстановить метаболизм липидов.
- Субстратредуцирующая терапия. Лечение направлено на снижение интенсивности образования патологически накапливаемого соединения (сложного липида, его метаболитов). Используются молекулы с низкой массой, которые стимулируют остаточную активность фермента.
- Симптоматическая терапия. Препараты подбираются индивидуально на основании клинической картины болезни. Распространено применение антиконвульсантов, обезболивающих средств, ингибиторов АПФ, гепатопротекторов. При отдельных видах липоидозов использование симптоматических лекарств является единственным способом лечения.
Прогноз и профилактика
Липоидозы характеризуются гетерогенностью генетического дефекта и выраженной клинической полиморфностью. Большинство из них имеет непрерывное прогрессирующее течение. Правильная диагностика и своевременное лечение позволяют увеличить продолжительность жизни больных, а при легких формах способствуют улучшению адаптации. Профилактика возможна на этапе планирования и в первые месяцы беременности. Супружеским парам с высоким риском передачи болезни ребенку рекомендуется медико-генетическое консультирование, а в первом триместре - исследование амниотической жидкости и материала биопсии хориона на наличие мутаций генов.
1. Лизосомные болезни накопления липидов у детей/ Захарова И.Н. и др.// Медицинский совет. - 2016 - №1.
Жировая болезнь печени - причины, симптомы, лечение и профилактика

Печень - самый крупный и важный орган брюшной полости. Обеспечивает белковый, углеводный и жировой обмен.
Жировая болезнь печени сегодня - очень распространенный недуг. Стереотип о том, что от ожирения печени страдают только люди с избыточным весом и алкоголики, вводит в заблуждение. Это заболевание вызывается не только чрезмерным употреблением алкоголя, но и нездоровым питанием и малоподвижным образом жизни, характерными для значительной части населения.
Средний возраст, в котором обнаруживают жировую дистрофию печени, составляет 50 лет, но в целом частота гепатоза увеличивается с возрастом: от 2,6% детей до 26% лиц в возрасте 40-50 лет.
Что такое жировая болезнь печени?
Здоровая печень должна содержать мало жира или вообще его не содержать. При жировой дистрофии или стеатозе печени жировые капли накапливаются в ее клетках - гепатоцитах.
Стеатоз печени развивается медленно, но даже на ранних стадиях может оказывать негативное влияние на сердечно-сосудистую систему. Также печень не способна достаточно эффективно очищать организм от лишнего холестерина и токсинов.
По мере того как количество поврежденных клеток возрастает до критического уровня, печень уже не в состоянии функционировать.
Жирная печень сама по себе не является большой проблемой. Но жировая ткань - это не инертное образование, она выделяет гормоны и способствует воспалению. Так что у некоторых людей — в среднем у 20% — развивается воспаление жировой печени или стеатогепатит, и это уже нехорошо.
У некоторых стеатогепатит прогрессирует и со временем вместо печеночных клеток развивается соединительная ткань и фиброз - путь к циррозу печени и развитию необратимых изменений в этом органе. Ткань печени замещается соединительной тканью и больше не функционирует. Следовательно, печень плохо выполняет свою работу - не производит, не обезвреживает, не очищает и не метаболизирует. Риск рака печени также выше.
Жировая дистрофия печени — серьезный диагноз, требующий наблюдения.
Поэтому очень важно следить за здоровьем печени и своевременно предотвращать причину заболевания, так как печень - единственный орган, способный восстановиться после развития очень тяжелого заболевания.
Печеночное ожирение чаще всего связано с двумя заболеваниями:
- Неалкогольная жировая болезнь печени или НАТАС;
- Алкогольная жировая болезнь печени или АТАС.
Причины возникновения жировой болезни печени
Почти каждому третьему человеку ставят диагноз жировая дистрофия или стеатоз, или жировой гепатоз печени после УЗИ брюшной полости. Больные с этими диагнозами часто не предъявляют жалоб, поэтому жировая дистрофия печени обнаруживается случайно.
Основные причины жировой дистрофии печени известны:
- Злоупотребление алкоголем. Печень расщепляет алкоголь, вырабатывает избыточную энергию, а организм откладывает ее в виде капелек жира.
- Слишком большое количество потребляемых человеком калорий - нездоровое питание, чрезмерное употребление насыщенных жирных кислот, например, сливочного масла и сахара. Особенно плохо так называемое центральное ожирение - в области живота скапливается лишний жир, потому что из кишечника и сальных оболочек в кровь поступает больше жирных кислот. Они попадают в печень и в ней накапливаются капельки жира.
- Один из метаболических синдромов: избыточный вес, диабет 2 типа, высокий уровень холестерина, гипертония или другие симптомы.
- Малоподвижный образ жизни.
- Нарушение функции инсулина. Инсулин — это гормон, способствующий синтезу белков и жиров в печени, он помогает снизить уровень сахара в крови после еды. В отсутствие инсулина неполное окисление промежуточных жиров в организме может привести к развитию сахарного диабета 2 типа.
- Вирусный гепатит В или С. Однако это случается редко.
- Иногда основная причина - регулярный прием лекарств. Около 99% применяемых лекарств метаболизируются непосредственно в печени. Поэтому потенциально любое лекарство может нанести ей вред. Исследования показывают, что антибиотики и нестероидные противовоспалительные препараты, статины, лекарства, используемые для лечения туберкулеза, грибковых заболеваний и психических заболеваний, с большей вероятностью вызывают повреждение печени.
Неалкогольная жировая болезнь печени
Неалкогольная жировая болезнь печени или НАТАС характеризуется избыточным накоплением жира в печени, что чаще всего встречается у пациентов с избыточным весом и диабетом, но также наблюдается у людей с нормальным весом, особенно у тех, кто не питается регулярно или недостаточно и склонны голодать. Худой трезвенник, но с жирной печенью? К сожалению, никто не застрахован от этого диагноза, потому что это болезнь образа жизни!
Алкогольная жировая болезнь печени
Алкоголь - причина жировой дистрофии печени, особенно если он употребляется регулярно. Однако если прием алкоголя прекратить, то жировая болезнь печени обычно исчезает. В этом существенное различие между алкогольной жировой болезнью печени и неалкогольной жировой болезнью печени.
Способность алкоголя воздействовать на печень во многом определяется дозой вещества. Небольшая доза - не более 10 граммов в день красного вина - даже укрепляет печень. Печень не повреждается и более высокой дозой алкоголя, но она не должна превышать 30 граммов в день. Но некоторые люди очень чувствительны к алкоголю, поэтому у них он может вызвать повреждение печени, даже если они употребляют его очень мало, но каждый день.
Быстрее всего жировая дистрофия печени образуется при регулярном употреблении алкоголя:
1) Для мужчин, если ежедневно выпивается 60 г алкоголя, то есть около 1,5 л пива или 0,6 л вина, или 120 г рома.
2) Для женщин, если ежедневно выпивать 20-40 г алкоголя, то есть примерно 0,5-1,0 л пива или 0,2-0,4 л вина.
Диагностика жировой болезни печени
Печень не болит. Пациенты с ожирением печени обычно не испытывают никаких проблем со здоровьем. Имеются только общие расстройства - слабость, повышенная утомляемость, тяжесть или стеснение в правом верхнем подреберье живота, потому что печень становится больше при отложении жира. Потенциальные проблемы легко диагностировать, поэтому рекомендуется периодически проверять и заботиться о своей печени.
- УЗИ брюшной полости;
- Оценка сопутствующих заболеваний - сахарный диабет, гипертоническая болезнь и другие;
- Анализ крови, обращая особое внимание на ферменты печени - АЛТ, АСТ, ГТФ, следует учитывать и другие показания - глюкозу, холестерин, ЛПНП и ЛПВП.
У пациентов с НАСГ АЛТ часто выше, чем АСТ. Но у больных с АТАС уровень АСТ в крови вдвое превышает уровень АЛТ.
Признаки хронического употребления алкоголя:
- кожные звездочки чаще всего поражают кожу лица;
- массивное расширение сосудов кожи и слизистых оболочек;
- эритема запястья - розовые круглые пятна на коже рук;
- гинекомастия у мужчин - увеличение молочных желез;
- увеличение слюнных желез;
- потливость, тахикардия, тремор рук;
- увеличение объема эритроцитов;
- увеличение ГГТ - фермент, находящийся в печени;
- концентрация АСТ > чем АЛТ.
Лечение
Лечение жировой дистрофии печени основано на устранении ее причин. Если больной употребляет алкоголь, его следует прекратить, а если жировая дистрофия печени обусловлена метаболическим синдромом, то следует контролировать уровень сахара в крови, уровень холестерина и снижать вес.
Основное лечение заключается в уменьшении избыточного жира. Пациентам с НАТАС и ожирением рекомендуется снижение массы тела на 7-10%. Этого можно добиться, сократив потребление калорий, увеличив физическую активность как минимум до двух-трех раз в неделю.
Несколько советов по снижению калорийности пищи:
- количество животных жиров и сахара в рационе должно быть ограничено;
- отказаться от колбас и жирных сортов мяса;
- научиться употреблять несладкий чай или кофе;
- не употреблять молоко и молочные продукты с повышенной или нормальной жирностью;
- ограничить употребление тортов и сладостей;
- не употреблять сладкие напитки;
- не употреблять алкоголь;
- печени понравится средиземноморская диета, основанная на растительных продуктах, рыбе, орехах и оливковом масле.
Если человек не может принять эти меры, то необходим прием лекарств для снижения веса - орлистата или сибутрамина. Если такое лечение не помогает, следует прекратить прием препарата и рассмотреть вопрос об операции по уменьшению жира.
Применение гепатопротекторов - эссенциальные фосфолипиды и силимарин - может способствовать очищению печени и улучшению ее биохимических свойств. Эссенциальные фосфолипиды обладают антиоксидантным и антифибротическим действием. Силимарин - экстракт облепихи. Исследования показали, что силимарин снижает резистентность к инсулину и может привести к снижению и нормализации АСТ и АЛТ.
Профилактика
Здоровая печень любит полноценное питание, и важно, чтобы оно было разнообразным. А именно чтобы клетки печени активно функционировали, им нужны как хорошие ненасыщенные жирные кислоты, так и углеводы для их переработки. Напротив, если человек ест однородную пищу, например, сидит на капустной или рисовой диете, часть клеток печени ленивы. Печень будет «наслаждаться» несколькими чашками кофе в день — доказано, что кофе снижает риск развития фиброза печени.
Чтобы избавиться от жира в печени, не обойтись без регулярных физических нагрузок, желательно с увеличением частоты сердечных сокращений. Подходит как для ходьбы, так и для медленного бега, езды на велосипеде, плавания, гребли, рекомендуются регулярные активные прогулки три-пять раз в неделю не менее получаса.
Даже если вес не уменьшился, жировая масса уменьшится в результате физической активности. Жиры — это триглицериды, которые расщепляются и уходят из печени в мышцы, потому что им нужна энергия для движения. У физически активного человека шансы заболеть стеатогепатитом становятся намного ниже.
Чтобы сохранить здоровье печени, необходимо вести здоровый образ жизни и стараться максимально отказаться и сократить вредные привычки. Своевременная борьба с причинами заболевания - лучшее решение. Профилактика определенно лучше, чем лечение.
Липидозы: причины, симптомы, диагностика, лечение
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Липидозы относятся к болезням накопления (тезаурисмозам), почти всегда протекают с поражением ЦНС, поэтому их называют нейролипидозами. Кожные проявления являются одним из основных симптомов только при диффузной ангиокератоме Фабри (гликосфинголипидоз), при других формах они возникают нечасто, возможно, из-за раннего летального исхода.
Гликоцереброзидоз (болезнь Гоше) - заболевание, в основе которого лежит снижение активности бета-глюкозидазы, накапливаются гликоцереброзиды в макрофагах (клетки Гоше) селезенки, костного мозга, лимфатических узлов, печени, легких, эндокринных желез, нейронах головного мозга и интрамуральных вегетативных ганглиев. Заболевание наследуется по аутосомно-рецессивному типу. Выделяют три клинических варианта: I - хронический, II - острый нейропатический (ювенильный). III - хронический нейропатический, которые, вероятно, обусловлены разными мутациями. Основными симптомами являются гепатоспленомегалия, церебральные дисфункции с судорогами, умственная отсталость, поражение костей. В коже может наблюдаться очаговая или диффузная пигментация, преимущественно открытых частей тела, при спленомегалии - петехии и экхимозы. Гистологически обнаруживают повышение содержаний меланина в эпидермисе, иногда и в верхней части дермы.
Болезнь Ниманна-Пика характеризуется накоплением фосфолипида сфингомиелина в нейронах и глиальных клетках головного мозга и элементах макрофагально-гистиоцитарной системы внутренних органов. Заболевание обусловлено дефектом активности сфингомиелиназы, наследуется по аутосомно рецессивному типу. Кожа вследствие повышения содержания меланина желтовато-коричневого цвета, могут наблюдаться ксантомы. Гистологически в некоторых случаях увеличивается количество пигмента в эпидермисе и определяются отдельные ксантомные клетки в дерме.
Ангиокератома тела диффузная (болезнь Андерсона-Фабри) обусловлена недостаточностью альфа-галактозидазы. W. Epinette и соавт. (1973) связывают ее с уменьшением активности а-L-фукозидазы. Описаны случаи болезни Фабри с нормальной активностью фермента, что свидетельствует о генетической неоднородности заболевания. Наследуется рецессивно, сцепленно с Х-хромосомой. В основе клинических проявлений лежит отложение липидов в эндотелиоцитах и перицитах кровеносных сосудов, гладких мышечных клетках, клетках ганглиев, нервах, эпителии роговицы, почек, кожи. Поражение кожи характеризуется появлением в детском или подростковом возрасте многочисленных мелких (диаметром 1-2 мм) темно-красных ангиокератом, преимущественно на нижней части туловиша, половых органах, бедрах, ягодицах, почти исключительно у мужчин. У женщин-гетерозигот могут быть изменения почек и глаз, очень редко кожи. Прогноз неблагоприятный, так как в среднем возрасте (40 лет) развиваются почечная недостаточность, инфаркт миокарда или инсульт.
Патоморфология. Обнаруживают резкое расширение капилляров сосочкового слоя кожи, стенки которых образованы одним слоем эндотелиоцитов, окруженного рыхлыми тяжами соединительной ткани. Эпидермальные отростки и волосяные фолликулы подвергаются атрофии от давления. Расширенные и переполненные кровью капилляры иногда близко прилежат друг к другу, образуя многокамерные полости, между которыми можно видеть длинные узкие эпидермальные выросты.
Верхняя часть подвергается атрофии, иногда отмечается незначительная вакуолизация клеток базального слоя эпидермиса. Гиперкератоз нередко бывает очень сильным, с явлениями паракератоза, особенно при поражении капилляров не только сосочкового, но и сетчатого слоев дермы. Липиды обнаруживают с помощью специфических методов окраски.
Необходима специальная фиксация биоптатов кожи в 1 % растворе хлорида кальция с добавлением 10% формалина или препараты содержат в 10 % формалине 2 дня, затем в 3 % растворе бихромата калия до 1 нед. После фиксации окрашивают по методу Тарновского. Липиды хорошо выявляются суданом черным Б и шарлахом. Они двоякопреломляющие, потому видны в поляризационном микроскопе.
Липиды находят не только в ангиоматозно измененных сосудах, но и в клинически неизмененной коже, в фибробластах, мышцах, поднимающих волос. Отложение липидов обнаруживают при электронной микроскопии в эндотелиоцитах, перицитах, фибробластах внутри крупных лизосом в виде двух видов внутриклеточных включений, окруженных двухконтурной мембраной и имеющих ламеллярную структуру. Лизосомы состоят из чередующихся между собой электронно-плотных и светлых полос. В большинстве из них выявляют активность фосфатаз, а в резидуальных телах находят миелиновые структуры.
Читайте также:
- УЗИ при расширении протока поджелудочной железы
- Врожденный вывих бедра и дисплазия тазобедренного сустава: атлас фотографий
- Спонтанная ( особая ) стенокардия ( стенокардия Принцметала ). Инфаркт миокарда. Клиника ( клиническая картина ) инфаркта миокарда. ЭКГ при инфаркте миокарда.
- КТ, МРТ при вертикальном продольном разрыве мениска коленного сустава
- Синдром Каплана-Кляцкина (Kaplan-Klatskin)