Синдром Улльриха-Фейхтигера (Ullrich-Feichtiger) - синонимы, авторы, клиника
Добавил пользователь Валентин П. Обновлено: 01.02.2026
Врождённые (конгенитальные) миопатии — группа генетически обусловленных миопатий, характеризующихся ранним началом (обычно с рождения до 1 года), характерным симптомокомплексом (синдромом «вялого ребёнка» ) и непрогрессирующим или медленно прогрессирующим течением. Среди конгенитальных миопатий выделяют конгенитальные мышечные дистрофии и конгенитальные миопатии (структурные миопатии).
КЛИНИЧЕСКАЯ КАРТИНА
Большинство конгенитальных миопатий и дистрофий проявляется синдромом «вялого ребёнка». Клинические проявления синдрома «вялого ребёнка» сводятся к выраженной мышечной гипотонии, слабому сопротивлению мышц при пассивных движениях, недержанию головы, задержке моторного развития, гипермобильности суставов. Синдром «вялого ребёнка» также наблюдают при спинальных мышечных атрофиях и других врождённых заболеваниях ЦНС, болезнях обмена (гликогенозы, аминоацидурии) и др. Приблизительно 80% случаев синдрома «вялого ребёнка» обусловлены первичным поражением ЦНС. Основной метод диагностики врождённых миопатий — морфологическое исследование мышц; ЭМГ лишь подтверждает первично-мышечный характер поражения на начальном этапе дифференциальной диагностики. Наряду с синдромом «вялого ребёнка» отмечаютслабость лицевой мускулатуры, мышц туловища и дыхательной мускулатуры. У большинства развиваются Koнтpaктypы тазобедренных, коленных и локтевых суставов, мышц-разгибателей шеи, сколиоз, позже может присоединиться наружная офтальмоплегия. Несмотря на задержку моторного развития, большинство детей способны самостоятельно сидеть, некоторые могут самостоятельно ходить (при частичном дефиците мерозина). При ЭМГ выявляют первично-мышечный тип изменений, при этом спонтанная активность мышечных волокон либо отсутствует, либо незначительна.
СИМПТОМЫ
Для конгенитальной мышечной дистрофии Фукуямы типичны выраженная мышечная слабость (дети либо вообще не способны самостоятельно ходить, либо начинают ходить только в возрасте 2-8 лет), симптоматическая эпилепсия (у 50%), умственная отсталость, офтальмологическая патология (микрофтальмия, гипоплазия сетчатки, катаракта, близорукость, косоглазие) . Характерны множественные и разнообразные изменения на МРТ (дисплазия, агирия, расширение желудочков, кисты). Для конгенитальной мышечной дистрофии Ульриха, помимо синдрома “вялого ребёнка”, характерны кифоз, гипермобильность дистальных суставов, дисплазия тазобедренных суставов, гиперкератоз. Способность к самостоятельной ходьбе зависит от тяжести течения, однако к 2- 10 годам, как правило, утрачивается из-за выраженных контрактур. Синдром Уолкера-Варбурга — одно из самых тяжёлых конгенитальных нервномышечных заболеваний (средняя продолжительность жизни — 9 мес). Наблюдают многочисленные врождённые аномалии: менингоцеле, агирию, агенезию мозолистого тела, гипоплазию пирамидного тракта, расширение желудочков, микроцефалию, микрофтальмию, гипоплазию зрительных нервов, катаракту, глаукому и другие изменения, приводящие к слепоте, синдром «вялого ребёнка» и бульбарные нарушения.
ЛЕЧЕНИЕ
Лечение должно производиться исключительно врачом-неврологом. Самолечение недопустимо. Специфической терапии не существует, цель лечения — коррекция ортопедических нарушений (дисплазия тазобедренных суставов, сколиоз), профилактика контрактур, поддержание мышечной силы, терапия кардиомиопатии и симптоматической эпилепсии.
Синдром Ульриха-Фейхтигера

Клиническая картина. При данном лор синдроме наблюдается аномалия развития наружного уха. Раннее снижение слуха обусловлено пороком развития внутреннего уха.
Со стороны лицевого скелета отмечается седловидная деформация носа, лицо имеет «восковой» (безэмоциональный) вид, за счёт нарушения иннервации мимической мускулатуры. Лобные кости увеличены, за счёт чего лоб имеет выпуклый вид. Рост у таких пациентов, как правило, выше среднего. Возможно расщепление твёрдого и мягкого нёба и верхней челюсти (заячья губа).
Со стороны органа зрения отмечается помутнение роговицы, катаракта глаза. Глазные щели сужены и уменьшены в размере (микроофтальмия). Реже данный порок сопровождается пороком сердца и поликистозом почечно-лоханочной системы.
Диагностика. Консультация лор врача, отомикроскопическое исследование, акуметрическое исследование слуха, аудиометрическое исследование слуха, исследование вызванными потенциалами мозга (в детском возрасте), тимпанометрическое исследование, исследование в позе Ромберга, воздушная нистагмометрия, консультация отоневролога, консультация медицинского генетика, консультация уролога, консультация нефролога, ультразвуковое исследование почек, общий анализ мочи, анализ мочи по Земницкому, экскреторное урографическое исследование, магнитно-резонансное томографическое исследование головного мозга, компьютерное томографическое исследование околоносовых пазух и височных костей, консультация офтальмолога, определение остроты зрения и осмотр глазного дна, консультация кардиолога, электрокардиографическое исследование.
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Врожденная мышечная дистрофия Ульриха - Ullrich congenital muscular dystrophy

врожденная мышечная дистрофия Ульриха - это форма врожденной мышечной дистрофии. Это связано с вариантами типа VI коллаген, обычно это связано с мышечной слабостью и респираторными проблемами, хотя проблемы с сердцем не связаны с этим типом CMD. Он назван в честь Отто Ульриха, который также известен своим синдромом Ульриха-Тернера.
Содержание
- 1 Признаки и симптомы
- 2 Генетика
- 3 Диагноз
- 3.1 Дифференциальный диагноз
- 4.1 Прогноз
Признаки и симптомы
Презентация Врожденная мышечная дистрофия Ульриха у пострадавшего выглядит следующим образом:
- слабость
- Затруднения при ходьбе (преимущественно проксимальных мышц, например шеи)
- Слабость в суставах (преимущественно в дистальных суставах)
Генетика
С точки зрения генетики врожденной мышечной дистрофии Ульриха, есть мутации в генах COL6A1, COL6A2, и COL6A3. Этот подтип мышечной дистрофии имеет аутосомно-рецессивный характер.
COL6A1 играет важную роль в поддержании целостности различных тканей человеческого тела. Субъединица альфа 1 типа VI коллаген представляет собой кодируемый белок.
Диагноз
С точки зрения диагностики врожденной мышечной дистрофии Ульриха при осмотре фолликулярного гиперкератоза, может быть дерматологическим индикатором, кроме того, уровень креатинкиназы сыворотки может быть несколько выше нормы. Другие исследования / методы для определения наличия у человека врожденной мышечной дистрофии Ульриха:
Дифференциальный диагноз
Лечение
Лечение врожденной мышечной дистрофии Ульриха может включать физиотерапия и регулярная растяжка для предотвращения и уменьшения контрактур. В какой-то момент пациенту может потребоваться респираторная поддержка.
Хотя сердечные осложнения не являются проблемой при этом типе ВМД, в отношении респираторных проблем возможна вентиляция через трахеостомию в некоторых случаях.
Прогноз
Прогноз этого подтипа MD указывает на то, что пострадавший человек может в конечном итоге иметь трудности с кормлением. В какой-то момент хирургическое вмешательство может быть вариантом лечения сколиоза.
Сколиоз, который представляет собой искривление позвоночного человека в сторону, определяется множеством факторов, включая степень (легкая или тяжелая), и в этом случае если возможно, человек может использовать бандаж.
Исследование
Что касается возможного исследования врожденной мышечной дистрофии Ульриха, один источник указывает, что циклоспорин A может быть полезным для людей с этим типом CMD.
Согласно обзору Bernardi, et al., циклоспорин A (CsA), используемый для лечения мышечной дистрофии коллагена VI, демонстрирует нормализацию митохондриальной реакции на ротенон..
Врожденные мышечные дистрофии
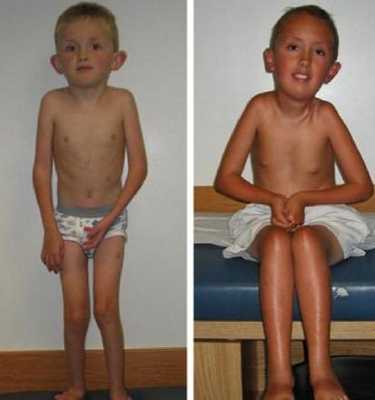
Это группа медленно прогрессирующих и непрогрессирующих клинически и генетически гетерогенных заболеваний, которые наследуются преимущественно по аутосомно-рецессивному типу. Врожденные мышечные дистрофии (ВМД) проявляются с рождения или в первые 12 месяцев жизни. Они сопровождаются гипотонией и прогрессирующей слабостью мышц, задержкой двигательного развития. Часто на фоне ВМД развиваются контрактуры, нарушения со стороны органов зрения и ЦНС.
Причины и формы врожденных мышечных дистрофий
Данные о молекулярно-генетических основах ВМД позволили составить классификацию патологий. Исходя из генетического и биохимического дефекта, выделяют такие группы врожденных мышечных дистрофий:
- обусловленные дефектами структурных протеинов (врожденная мышечная дистрофия Ульриха, Бетлема, мерозин-дефицитная форма, интегрин-дефицитная, с буллезным эпидермолизом, с гипермобильностью суставов);
- связанные с дефектами гликозилирования (синдром Уокера-Варбурга, цереброокулярная миодистрофия, ВМД Фукуямы, врожденные мышечные дистрофии с гипертрофией мышц, с аномальным гликозилированием альфа-дистрогликана и тяжелой умственной отсталостью, с псевдогипертрофией, макроглоссией и дыхательной недостаточностью);
- вызванные дефектами белков эндоплазматического ретикулума и ядра (синдром ригидного позвоночника, LMNA-дефицитная врожденная мышечная дистрофия);
- прочие формы (ВМД с нарушением структуры митохондрий и т. д.).
Признаки врожденной мышечной дистрофии
При рождении обнаруживается выраженная слабость и гипотония мышц. Наблюдается слабость лицевых мышц, дисфагия, нарушения дыхательной функции, контрактуры суставов (с момента рождения или развивающиеся со временем, в т. ч. врожденный множественный артрогрипоз — контрактуры двух и более суставов). ВМД сопровождаются врожденными вывихами бедер (25 % случаев), деформациями позвоночника.
Интеллект обычно не страдает. Исключение составляют такие формы врожденной мышечной дистрофии, как ВМД Фукуямы и синдром Уокера-Варбурга. Для первой формы характерно наличие аномалий извилин коры большого мозга и мозжечка, глубокое слабоумие, фебрильные и афебрильные судороги, повышенный уровень КФК в крови. При синдроме Уокера-Варбурга помимо аномалий извилин мозга присутствуют гидроцефалия и патологии глаз.
Диагностика врожденных мышечных дистрофий
Исследование включает в себя:
- электромиографию;
- анализ на КФК;
- биопсию мышц;
- МРТ головного мозга.
Подтвердить диагноз позволяют молекулярно-генетические тесты, направленные на поиск мутаций в соответствующих генах. За дефекты структурных протеинов отвечают такие гены, как LAMA2, COL6A1, COL6A2, COL6A3, ITGA7, ITGA9, PLEC. Дефекты гликозилирования связаны с генами: POMT1, POMT2, FCMD, FKRP, LARGE, POMGnT1, FKRP. Установлена связь дефектов белков эндоплазматического ретикулума и ядра с нарушениями в генах: N1, SEPN1, SBP2, LMNA. Прочие формы ВМД обусловлены поломками в гене CHKB и др.
В медико-генетическом центре «Геномед» проводятся исследования, направленные на поиск мутаций в вышеперечисленных генах, которые могут оказаться причиной той или иной формы врожденной мышечной дистрофии.
Терапия врожденных мышечных дистрофий преимущественно поддерживающая. Для профилактики контрактур и поддержания силы мышц больным показана лечебная физкультура. Хирургическое вмешательство оправдано при серьезных деформациях позвоночника.
Синдром Криглера-Найяра
Синдром Криглера-Найяра - генетическое заболевание из класса ферментопатий, характеризующееся нарушением одного из звеньев процесса обезвреживания и выведения билирубина - конъюгации. Симптомами этого состояния являются желтуха печеночного генеза и тяжелые неврологические нарушения, которые могут привести к летальному исходу еще в младенческом возрасте. Диагностика синдрома Криглера-Найяра производится посредством биохимических проб и определения уровня неконъюгированного билирубина в плазме крови, а также молекулярно-генетическими методиками. Специфического лечения заболевания не существует (за исключением трансплантации печени), терапия сводится к увеличению разрушения и элиминации билирубина из организма (гемосорбция, фототерапия, плазмаферез, прием барбитуратов).
Общие сведения
Синдром Криглера-Найяра - тяжелое генетическое заболевание, характеризующееся нарушением связывания билирубина с глюкуроновой кислотой, что является ключевым этапом его обезвреживания и выведения из организма. Впервые это заболевание было описано в 1952 году двумя американскими педиатрами - Джоном Криглером и Виктором Найяром. Дальнейшее изучение синдрома Криглера-Найяра показало, что это состояние имеет генетическую природу и аутосомно-рецессивный характер наследования, кроме того, удалось выявить две клинические разновидности данной патологии. Заболевание достаточно редкое, поэтому точные цифры встречаемости не определены - большинство исследователей полагает, что она находится на уровне 1:1 000 000. Половое распределение больных синдромом Криглера-Найяра не имеет особенностей, заболевание с одинаковой частотой поражает как мальчиков, так и девочек. В лечении этого состояния крайне важна ранняя (в идеале - пренатальная) диагностика, так как от своевременности начатой терапии очень сильно зависят прогноз заболевания и качество жизни больного.
Причины
Синдром Криглера-Найяра относят к классу ферментопатий (по другой классификации - к группе неконъюгированных гипербилирубинемий), причина этого заболевания кроется в недостаточности уридиндифосфатглюкуронидазы 1, функцией которой является связывание билирубина с двумя молекулами глюкуроновой кислоты. В итоге этого биохимического процесса билирубин становится способным растворяться в воде, выводиться в составе желчи и, главное, значительно падает его токсичность. При синдроме Криглера-Найяра этот процесс резко замедлен или не происходит совсем, вследствие чего возникает задержка элиминации билирубина из организма и его накопление.
Билирубин обладает выраженной нейротоксичностью, при повышении концентрации в крови это вещество начинает откладываться в тканях кожных покровов и слизистых оболочек, приводя к развитию желтухи. Когда концентрация билирубина превышает определенный порог, соединение начинает проникать через гематоэнцефалический барьер в головной мозг, приводя к тяжелой энцефалопатии (особенно повреждаются базальные ядра). При отсутствии лечения больные синдромом Криглера-Найяра погибают от многочисленных неврологических расстройств и нарастающей печеночной комы.
Причиной низкой активности уридиндифосфатглюкуронидазы являются мутации гена UGT1A1, который располагается на 2-й хромосоме, отвечает за аминокислотную последовательность и выделение этого фермента. Помимо синдрома Криглера-Найяра дефекты этого гена могут приводить к ряду других нарушений билирубинового обмена наследственного характера - синдрому Жильбера, транзиторной неонатальной билирубинемии семейного типа. Механизм наследования мутаций гена UGT1A1 при синдроме Криглера-Найяра аутосомно-рецессивный. При этом описано несколько вариантов возможного повреждения этого гена, которые приводят к разному течению данного заболевания.
Классификация и симптомы синдрома Криглера-Найяра
В настоящее время описаны две основные клинические формы синдрома Криглера-Найяра, в основном различающиеся между собой тяжестью проявлений и прогнозом заболевания. Это обусловлено типом генетического дефекта в UGT1A1. Первый тип заболевания (СКН-1) вызывается миссенс-мутациями, приводящими к появлению неполноценного фермента, имеющего сигнальную последовательность аминокислот, характерную для подвергающихся внутриклеточной утилизации белков. Таким образом, при этой форме дефект гена поражает кодирующие участки (экзоны), что вызывает развитие патологии у гомозигот. Вскоре после своего образования уридиндифосфатглюкуронидаза 1 разрушается и конъюгации билирубина не происходит совсем.
Синдром Криглера-Найяра 1-го типа характеризуется тяжелым и стремительным течением - первые признаки гипербилирубинемии в виде желтухи обнаруживаются уже через несколько часов после рождения. Со временем к ним присоединяются неврологические нарушения - нистагм, судорожные приступы, иногда возникает опистотонус. Желтуха сохраняется на протяжении всей жизни ребенка, его умственное развитие резко отстает от такового у сверстников, симптомы заболевания неуклонно нарастают даже при интенсивном лечении. Обычно больные синдромом Криглера-Найяра 1-го типа умирают на протяжении первого года жизни из-за интоксикации билирубином и поражения базальных подкорковых ядер (ядерная энцефалопатия).
Причиной синдрома Криглера-Найяра 2-го типа также являются миссенс-мутации гена UGT1A1, однако они могут возникать как в кодирующей последовательности (экзонах), так и в промоторе - участке, отвечающем за экспрессию данного гена. У большинства больных СКН-2 наблюдается наличие на одной хромосоме дефекта экзона, на другой - промотора, то есть, такие лица являются компаунд-гетерозиготами. Результатом нарушения является продукция дефектной формы фермента уридиндифосфатглюкуронидазы, которая не разрушается, но имеет пониженную (на уровне 20-25% от нормы) функциональную активность. Поэтому синдром Криглера-Найяра 2-го типа характеризуется менее тяжелой клинической картиной и более благоприятным прогнозом.
В первые месяцы и даже годы жизни больных синдром Криглера-Найяра этого типа нередко проявляется только незначительной желтухой, при отсутствии лечения к подростковому периоду могут развиваться неврологические отклонения. В ряде случаев, особенно при правильно назначенных терапевтических мероприятиях, никаких нарушений со стороны центральной нервной системы не возникает вовсе. Проявления желтухи различной степени выраженности у больных синдромом Криглера-Найяра 2-го типа могут сохраняться на протяжении всей жизни и нередко расцениваются как индикатор осложнений и ухудшения состояния пациента. С возрастом иногда появляется нистагм, могут регистрироваться судорожные припадки, однако течение и выраженность симптомов заболевания всецело зависят от качества лечения и выполнения рекомендаций специалистов.
Диагностика
Диагностика синдрома Криглера-Найяра производится на основании данных общего осмотра ребенка, биохимических исследований крови, желчи и мочи, молекулярно-генетических анализов. При осмотре выявляется желтуха, возникшая в первые часы (при СКН-1) или месяцы (СКН-2) жизни, признаки неврологических нарушений (опистотонус, нистагм, длительное сохранение транзиторных рефлексов). У больных 2-м типом синдрома Криглера-Найяра неврологические расстройства могут регистрироваться во взрослом возрасте, тогда как у детей наблюдается только желтуха. Также с возрастом могут присоединяться такие проявления, как нейросенсорная глухота или хореоатетоз.
При биохимическом исследовании крови выявляется выраженная непрямая гипербилирубинемия (вплоть 200-350 мкмоль/л), отсутствие (при синдроме Криглера-Найяра 1-го типа) или резкое снижение концентрации прямого билирубина. Конъюгированная фракция этого соединения отсутствует в желчи при СКН-1 и присутствует в незначительных количествах при СКН-2. Фенобарбиталовая проба при синдроме Криглера-Найяра положительна только в случае наличия уридиндифосфатглюкуронидазы, то есть при СКН-2. Изучение концентрации неконъюгированного билирубина в моче показывает его увеличение. Молекулярно-генетическая диагностика синдрома Криглера-Найяра производится врачом-генетиком - он совершает прямое секвенирование последовательности гена UGT1A1 с целью выявления мутаций. При отягощенной по этому заболеванию наследственности у родителей может осуществляться пренатальная диагностика патологии. Дифференциальный диагноз следует проводить с обычной транзиторной желтухой новорожденных и синдромом Жильбера.
Лечение синдрома Криглера-Найяра
Специфического или этиотропного лечения синдрома Криглера-Найяра на сегодняшний день не существует, все терапевтические мероприятия назначаются для ускорения распада билирубина, его выведения из организма и защиты ЦНС. Особых отличий в терапии 1-го или 2-го типа заболевания нет (за исключением активизации микросомального окисления барбитуратами, которая не производится при 1-м типе), однако при СКН-1 лечение лишь незначительно оттягивает наступление летального исхода. Самым радикальным методом лечения синдрома Криглера-Найяра в настоящее время является операция по аллотрансплантации печени от родственника или генетически сходного донора - в этом органе происходит образование уридиндифосфатглюкуронидазы.
Синдром Криглера-Найяра 2-го типа лечат назначением умеренных доз барбитуратов для активации окисления билирубина и увеличения образования нужного фермента. Кроме того, показаны плазмаферез, гемосорбция, заместительное переливание крови - все эти процедуры направлены на удаление неконъюгированного билирубина из организма. Неплохие результаты у больных синдромом Криглера-Найяра дает фототерапия - облучение кожных покровов приводит к частичному разрушению билирубина и освобождению рецепторов тканей для новых порций этого токсина, что снижает его концентрацию в крови. Правильный питьевой режим и повышенное потребление жидкости ускоряет выведение токсина из организма, поэтому следует избегать обезвоживания. Необходим постоянный мониторинг уровня этого вещества в плазме крови, особенно опасным считается его количество свыше 300-340 мкмоль/л - при такой концентрации билирубин становится способным проникать через гематоэнцефалический барьер.
Прогноз и профилактика
Прогноз синдрома Криглера-Найяра 1-го типа исключительно плохой - из-за полного отсутствия активности фермента уридиндифосфатглюкуронидазы 1 больные умирают на протяжении первого года жизни из-за осложнений ядерной энцефалопатии. Течение СКН-2 зависит от таких факторов, как выраженность проявлений, своевременность диагностики и начала лечения, соблюдения рекомендаций специалистов, наличия или отсутствия сопутствующих заболеваний. В большинстве случаев прогноз относительно благоприятный - больные синдромом Криглера-Найяра 2-го типа могут прожить до преклонного возраста, из характерных проявлений патологии их может беспокоить только желтуха. Профилактика этого состояния возможна только в рамках консультации генетика для родителей, имеющих отягощенную наследственность по этому заболеванию, а также при помощи пренатальной диагностики.
Читайте также: