Виды наследственных полиневропатий. Амиотрофии. Лечение наследственных полиневропатий.
Добавил пользователь Alex Обновлено: 21.01.2026
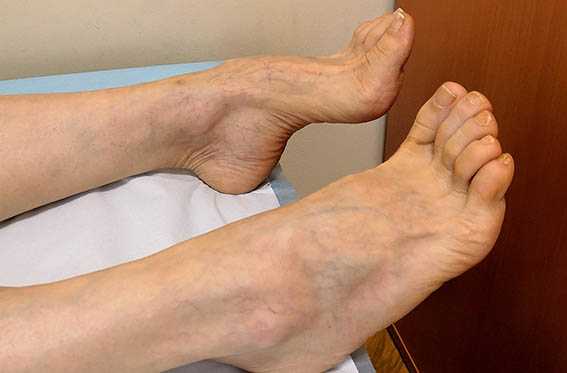
Полинейропатия (ПНП) (информация для пациента)
Полинейропатия - одна из самых распространенных болезней периферической нервной системы.
Что такое полинейропатия?
Полинейропатии - это группа заболеваний, характеризующаяся множественным и диффузным поражением корешков, сплетений и периферических нервов.
- чувствительные симптомы: пациента беспокоят онемение, покалывание, жжение, «ползанье мурашек» в кистях и/или стопах; неустойчивость при ходьбе, усиливающаяся в темноте и при закрывании глаз;
- двигательные симптомы: развивается неловкость, слабость в кистях и/или стопах; похудание мышц рук и ног; нарушение мелкой моторики (сложности при застегивании пуговиц и молнии, завязывании шнурков и т.д.); затруднения при ходьбе («шлепанье» стоп, трудности при подъеме и спуске с лестницы и т.д.);
- вегетативные симптомы: колебание цифр артериального давления, «перебои» в работе сердца, запоры или диарея, сухость кожи или повышенная потливость, снижение либидо, нарушение эрекции.
- острые полинейропатии (развитие в течение < 4-х недель): наиболее частой причиной является синдром Гийена-Барре;
- подострые полинейропатии (развитие в течение 4-8 недель);
- хронические полинейропатии (развитие в течение > 8 недель): частыми причинами является сахарный диабет, злоупотребление алкоголем, недосток витаминов группы В, хроническая воспалительная демиелинизирующая полинейропатия, наследственные причины и др.
- соблюдение диеты, неправильное питание, вегетарианство, приводящие к развитию дефицита витаминов группы B12;
- проведение химиотерапии, использование нейротоксичных препаратов, реже - интоксикация тяжелыми металлами и др.;
- аутоиммунное поражение периферических нервов с развитием дизиммунных нейропатий (синдром Гийена-Барре, хроническая воспалительная демиелинизирующая полинейропатия, парапротеинемические полинейропатии, мультифокальная моторная нейропатия и др.);
- хронические соматические заболевания: системные аутоиммунные заболевания соединительной ткани (системная красная волчанка, ревматоидный артрит, болезнь Шегрена и др.), хроническая почечная или печеночная недостаточность, патология щитовидной железы и др.;
- инфекционные заболевания (Лайм-боррелиоз, ВИЧ-инфекция, нейросифилис);
- онкологические заболевания с развитием паранеопластического процесса;
- наследственные нейропатии (наследственные моторно-сенсорные нейропатии, наследственная нейропатия со склонностью к параличам от сдавления, транстиретиновая семейная амилоидная полинейропатия, порфирийная полинейропатия и др.).
- электронейромиография (ЭНМГ) - основной инструментальный метод диагностики болезней периферических нервов, который не только подтверждает факт поражения нервов, но и определяет характер их повреждения (демиелинизирующий, аксональный, с блоками проведения).
- комплексное лабораторное исследование крови: минимальный объем - развернутый общий и биохимический анализ крови, RW, анти-ВИЧ, HBsAg и анти-HCV, уровень витаминов В1, В6, В9, В12, гомоцистеин (при необходимости, в каждом случае индивидуально, спектр лабораторного обследования расширяется)
- при подозрении на наследственную нейропатию - молекулярно-генетический анализ (поиск мутаций в гене PMP22, панель «нервно-мышечные заболевания» или полное геномное секвенирование) и т.д.;
- электрофорез белков сыворотки и мочи с иммунофиксацией + freelite;
- люмбальная пункция с общим анализом ликвора;
- инструментальное общесоматическое обследование, в том числе онкоскрининг (КТ легких, УЗИ молочных желез, маммография, УЗИ простаты и мошонки, ЭГДС, колоноскопия, УЗИ органов брюшной полости и малого таза, ПЭТ-КТ и т.д.).
Какие методы лечения разработаны при полинейропатии?
С позиций доказательной медицины не для всех полинейропатий разработано патогенетическое лечение.
Так, при диабетической полинейропатии, прежде всего, необходим тщательный контроль уровня глюкозы крови и соблюдение всех рекомендаций лечащего эндокринолога.
При токсической, например, алкогольной полинейропатии, лечение начинается с полного отказа от вредной привычки.
При обнаружении дефицитарной полинейропатии проводят витаминотерапию, а также устраняют причины, вызывающие развитие заболевания.
Патогенетическая терапия (т.е. терапия, направленная на коррекцию механизмов развития заболевания) является основной в лечении дизиммунных полинейропатии (синдром Гийена-Барре, хроническая воспалительная демиелинизирующая полинейропатия, парапротеинемические полинейропатии, мультифокальная моторная нейропатия и др.). В зависимости от типа дизиммунной нейропатии, в индивидуальном порядке, учитывая все нюансы заболевания, подбирается оптимальный метод лечения или их сочетание. Разработаны следующие методы патогенетической терапии дизиммунных нейропатий: гормонотерапия (применение высоких доз глюкокортикостероидных препаратов), высокообъемный плазмаферез, высокодозная внутривенная иммунотерапия, а также применение цитостатиков и препаратов моноклональных антител.
- Транстиретиновая семейная амилоидная полинейропатия (ТТР-САП) - трансплантация печени, стабилизация молекулы транстиретина;
- Болезнь Фабри - фермент-заместительная терапия;
- Порфирийная полинейропатия - аргинат гема.
- Болезнь Рефсума - диетотерапия, плазмаферез.
Восстановительно-реабилитационные методы лечения (чрескожная электронейростимуляция, акупунктура, биологическая обратная связь, интервенционные методы, массаж, физиотерапия, баланстерапия, роботизированная терапия) занимает важное место в коррекции неврологических нарушений при полинейропатии.
Если у вас есть симптомы полинейропатии или вам поставлен диагноз "Полинейропатия", вы можете пройти комплексное обследование в центре заболеваний периферической нервной системы ФГБНУ НЦН, где вам помогут уточнить диагноз, выявить причины поражения периферических нервов и назначат терапию с позиций доказательной медицины.
Для ФГБНУ НЦН данное направление издавна является одним из приоритетных. Здесь было создано первое в стране отделение нейрореанимации, которое славилось уникальной методологией ухода за пациентами с тяжелыми формами полинейропатий, нуждающимися в длительной искусственной вентиляцией легких. Несколько десятилетий назад были проведены уникальные работы по диагностике и лечению отдельных форм нейропатий: наследственных, дифтерийной, дизиммунных и др. Здесь впервые в СССР был внедрён плазмаферез как метод лечения аутоиммунных заболеваний нервной системы. Накоплен уникальный отечественный опыт лечения и реабилитации больных с синдромом Гийена-Барре, тяжелыми формами ХВДП и нейропатий другого генеза.
В состав центра заболеваний периферической нервной системы входит 13 специалистов, из них 10 неврологов и 3 эндокринолога. Все неврологи владеют методиками ЭНМГ-исследования.
Сотрудники центра заболеваний периферической нервной системы консультируют пациентов амбулаторно в рамках ОМС и на коммерческой основе.
Наследственная моторная и сенсорная невропатия
Наследственные невропатии представляют собой гетерогенную группу генетически детерминированных заболеваний нервной системы, проявляющихся множественным поражением периферических нервов и различающихся типом наследования, выраженностью клинического полиморфизма, особенностями течения, характером электромионейрографических и морфогистохимических изменений.
Протокол "Наследственная моторная и сенсорная невропатия"
Коды по МКБ-10: G 60,0
- Болезнь Шарко-Мари Тута (наследственная мотосенсорная невропатия, тип I)
- Болезнь Дежерина - Сотта (наследственная мотосенсорная невропатия, тип III)
- Наследственная моторная и сенсорная невропатия, типы I-IV
- Гипертрофическая невропатия у детей
- Перонеальная мышечная атрофия (аксональный тип, гипертрофический тип)

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
Клинико-генетическая классификация наследственных мотосенсорных невропатий
Аутосомно-доминантная наследственная мотосенсорная невропатия, тип I:
- обусловленная дупликацией 17 р 11.2;
- обусловленная точковыми мутациями гена миелина РМР-22.
Тип I В: обусловленная точковыми мутациями гена миелина Ро.
Тип I С: с неидентифицированным генетическим дефектом.
Аутосомно-рецессивная наследственная мотосенсорная невропатия, тип I: с неидентифицированным генетическим дефектом.
Аутосомно-доминантная наследственная мотосенсорная невропатия, тип II: с неидентифицированным генетическим дефектом.
Аутосомно-рецессивная наследственная мотосенсорная невропатия, тип II: с неидентифицированным генетическим дефектом.
Наследственная мотосенсорная невропатия, тип III:
- обусловленная точковыми мутациями гена миелина РМР-22;
- обусловленная точковыми мутациями гена миелина Ро.
Х- сцепленная наследственная мотосенсорная невропатия:
- обусловленная точковыми мутациями и делециями коннексина 32;
- с неидентифицированным генетическим дефектом.
Сложные формы наследственных мотосенсорных невропатий:
- с пирамидными симптомами;
- с атрофией зрительных нервов;
- с атрофией зрительных нервов и глухотой;
- с пигментной ретинопатией;
- с кератодермией ладоней и стоп.
Диагностика
Диагностические критерии
Жалобы и анамнез: мышечная утомляемость, атрофии, боли в дистальных отделах конечностей по «полиневритическому типу», костные деформации, нарушение походки, задержка моторного развития, прогрессирующее течение.
В анамнезе: при болезни Шарко-Мари - аутосомно-рецессивный тип наследования, дебют заболевания в первое десятилетие жизни.
При болезни Дежерина-Сотта (наследственной мотосенсорной невропатии, тип III): аутосомно-рецессивный тип наследования, дебют в первые два года жизни, задержка моторного развития, слабость и атрофии дистальных отделов конечностей, по мере прогрессирования - поражение проксимальных отделов, атаксия, быстропрогрессирующее течение.
Для наследственной мотосенсорной невропатии, тип I характерен аутосомно-доминантный тип наследования, дебют заболевания преимущественно в 1-2 десятилетие жизни, медленно-прогрессирующий характер течения.
Для наследственной мотосенсорной невропатии, тип II - характерен аутосомно-доминантный тип наследования, дебют во второй декаде жизни, отсутствие либо незначительно выраженные атрофии дистальных отделов конечностей, относительно доброкачественное течение.
Для наследственной мотосенсорной невропатии, тип II - характерен аутосомно-рецессивный тип наследования, дебют заболевания в раннем возрасте, выраженные атрофии и слабость дистальных отделов конечностей, деформации кистей и стоп, быстропрогрессирующий характер течения.
Физикальное обследование: неврологический статус - боли (ноющие, стреляющие, крампи) в дистальных отделах конечностей на фоне гипостезии, с постепенным присоединением слабости, атрофии в дистальных отделах, снижение и утрата ахилловых рефлексов по мере прогрессирования - карпо-радиальных; нарушение походки по типу «петушиной», степпаж.
Атрофии возникают в дистальных отделах с последующей (при прогрессировании) атрофией проксимальных отделов конечностей. Деформированные стопы по типу «клюшки» или «фридрейховской» стопы в виде полой стопы с высоким сводом, экстензией основных фаланг. При дебюте заболевания в детском возрасте - задержка моторного развития.
Лабораторные исследования: общий анализ крови и мочи без патологии.
Инструментальные исследования
ЭМГ различна: для наследственной мотосенсорной невропатии, тип I характерно снижение скорости проведения импульса по моторным и сенсорным волокнам.
Для болезни Шарко-Мари снижение скорости проведения импульса по моторным и сенсорным волокнам.
Для болезни Дежерина-Сотта значительное снижение (менее 12 м/с) скорости проведения импульса по периферическим нервам.
Для наследственной мотосенсорной невропатии, тип II (аутосомно-рецессивная форма) на ЭМГ характерно значительное снижение скорости проведения импульса по периферическим нервам (менее 38 м/с).
Для наследственной мотосенсорной невропатии, тип II (аутосомно-доминантная форма) умеренное снижение скорости проведения импульса по периферическим нервам.
Показания для консультации специалистов:
Минимум обследования при направлении в стационар:
1. Общий анализ крови и мочи.
4. Кал на яйца глист.
Основные диагностические мероприятия:
1. Общий анализ крови.
2. Общий анализ мочи.
Дополнительные диагностические мероприятия:
Дифференциальный диагноз
Дифференциально-диагностические признаки аутосомно-доминантных наследственных мотосенсорных невропатий
Невральная амиотрофия Шарко-Мари-Тута
Невральная амиотрофия Шарко-Мари-Тута — это прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.
МКБ-10

Общие сведения
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания.
По различным данным, невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ. Лица мужского пола болеют чаще, чем женщины.
Причины
На сегодняшний день практическая неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования.
Патогенез
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение.
- Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса. Биопсия нерва обнаруживает сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток;
- При амиотрофии ШМТ II типа скорость проведения страдает незначительно, анализ биоптата показывает дегенерацию аксонов.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
Симптомы
Невральная амиотрофия Шарко-Мари-Тута начинается с развития симметричных мышечных атрофий в дистальных отделах ног. Начальные симптомы манифестируют, как правило, в первой половине второго десятилетия жизни, реже в период от 16 до 30 лет. Они заключаются в повышенной утомляемости стоп при необходимости длительно стоять на одном месте. При этом наблюдается симптом «топтания» - чтобы снять утомляемость стоп пациент прибегает к ходьбе на месте.
В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов. Основной симптом, на который пациенты чаще всего сами обращают внимание - приступообразные болезненные сокращения в икроножных мышцах (крампи), усиливающиеся в ночное время или после длительной физической нагрузки.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей. Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет. Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние. Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз.
Осложнения
Невральная амиотрофия Шарко-Мари-Тута характеризуется ранней инвалидизацией. Вследствие прогрессирующей атрофии дистальных отделов конечностей и выраженных нарушений чувствительности больные постепенно теряют способность к самостоятельной ходьбе. Из-за грубых деформаций кистей рук пациенты не могут сами себя обслуживать. Контрактуры суставов нередко требуют хирургической коррекции.
На ранней стадии заболевания слабость в мышцах ног, гипестезия и гипорефлексия приводят к частым падениям, что повышает вероятность травм и переломов. Наиболее грозные неблагоприятные последствия происходят при сочетании болезни Шарко-Мари-Тута и атаксии Фридрейха. К ним можно отнести слепоту, кардиомиопатию, дыхательную недостаточность.
Курацией пациентов занимаются врачи-неврологи и ортопеды. При опросе больного уточняется возраст, в котором начали появляться симптомы (для болезни ШМТ типична манифестация в 15-25 лет). Важное значение имеет семейный анамнез (наличие близкого родственника с этой патологией). Во время общего осмотра обращается внимание на изменение походки, деформацию стоп и кистей.
При неврологическом осмотре отмечается уменьшение тонуса дистальных отделов верхних и нижних конечностей, ослабление или полное отсутствие сухожильных рефлексов (ахилловых, коленных), снижение кожной чувствительности. Для уточнения диагноза проводятся следующие методы исследования:
Дифференциальный диагноз невральной амиотрофии Шарко-Мари-Тута необходимо проводить с наследственными нейромышечными заболеваниями (спинальная мышечная атрофия Верднига-Гоффмана, адренолейкодистрофия, болезнь Пелицеуса-Мерцбахера) и приобретенными хроническими полинейропатиями (синдром Гийена-Барре).
Лечение невральной амиотрофии Шарко-Мари-Тута
Медикаментозная терапия
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В настоящее время не существует специфической терапии, способной замедлить прогрессирование аксональной дегенерации и демиелинизации. Однако своевременно начатая грамотная и индивидуально подобранная терапия способна значительно улучшить качество жизни пациентов. Из лекарственных препаратов для симптоматического лечения невральной амиотрофии ШМТ применяются:
- Витамины. Для улучшения микроциркуляции и восстановления нервных волокон назначаются инъекции витаминов группы В (В1, В3, В12). К витамину В6 стоит относиться с осторожностью, так как превышение его дозы оказывает нейротоксический эффект. По данным некоторых исследователей, аскорбиновая кислота способна подавлять образование периферического белка миелина (PMP22).
- Миорелаксанты. С целью устранения болезненных мышечных сокращений пациентам рекомендуется прием медикаментов, расслабляющих скелетную мускулатуру - баклофен, толперизон.
- Кальций и витамин Д. Так как примерно 40% больных имеют остеопороз, для уменьшения риска переломов им показаны препараты кальция и витамина Д (холекальциферол).
- Антихолинэстеразные средства. При болезни ШМТ 2 типа для улучшения нервно-мышечной проводимости целесообразно назначение прозерина, галантамина.
Немедикаментозная терапия
Основное внимание уделяется немедикаментозному лечению невральной амиотрофии Шарко-Мари-Тута. Для достижения максимального терапевтического эффекта применяется комплекс следующих мероприятий:
- Электростимуляция. Для усиления нейротрофики, активации метаболизма в паретичных мышцах и проводимости периферических нервов используется направленная подача электрических импульсов.
- ЛФК. С целью повышения мышечного тонуса рекомендуются регулярные занятия лечебной физкультурой. Наиболее эффективно совмещение активных (выполняются самим пациентом) и пассивных (выполняются специалистом) упражнений.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах (в первую очередь нижних конечностей) выполняются различные виды массажа - ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Бальнеотерапия. Грязевые ванны и грязевые аппликации способствуют коррекции нарушений вегетативной нервной системы и замедлению формирования контрактур.
- Ортопедическое лечение. Чтобы предупредить развитие грубых деформаций больным назначается ношение ортопедической обуви. При нестабильности суставов из-за мышечной слабости, для фиксации стоп в заданном положении используются специальные приспособления (ортезы, подтяжки).
Хирургическое лечение
При выраженных атрофических явлениях и деформации стопы, значительно затрудняющих самостоятельную ходьбу, когда консервативные методы оказываются безуспешными, показаны ортопедические оперативные вмешательства - метатарзальная остеотомия, остеотомия пяточной кости. В некоторых случаях для восстановления опорной функции стопы может понадобиться проведение артродеза.
Экспериментальное лечение
Продолжаются поиски эффективного лекарства для борьбы с невральной амиотрофией Шарко-Мари-Тута. В клинических испытаниях, где пациенты принимали препарат PXT3003 (комбинация малых доз баклофена, налтрексона и сорбитола), были отмечены положительные результаты в виде увеличения мышечной силы, возобновления чувствительности и сухожильных рефлексов.
Рассматривается возможность использования в качестве лечения ингибиторов HDAC6 - ферментов, стимулирующих регенерацию белков цитоскелета нервных клеток. Эксперименты на лабораторных животных показали, что данные вещества способны значительно замедлить прогрессирование демиелинизации и аксональной дегенерации.
Прогноз и профилактика
Невральная амиотрофия Шарко-Мари-Тута - тяжелое инвалидизирующее заболевание. Большинство пациентов утрачивают способность ходить через 15-20 лет после начала появления симптомов. Однако в виду того, что преимущественно поражаются дистальные отделы конечностей, продолжительность жизни больных практически не отличается от таковой в общей популяции.
Летальные исходы в молодом и среднем возрасте наблюдаются при сочетании с атаксией Фридрейха, когда в патологический процесс вовлекается дыхательная мускулатура и миокард. Специфических методов первичной профилактики не существует. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
1. Оценка качества жизни больных с наследственной невропатией Шарко—Мари—Тута в Красноярском крае/ Шнайдер Н.А., Глущенко Е.В., Козулина Е.А.// Бюллетень Сибирской медицины. - 2011.
2. Клинико-генетическая характеристика наследственной невропатии Шарко—Мари—Тута (на примере Красноярского края): Автореферат диссертации/ Глущенко Е.В. - 2011.
3. Ведение и реабилитация пациентов с наследственной невропатией Шарко—Мари—Тута/ Шнайдер Н.А., Глущенко Е.В.// Комплексная реабилитация: наука и практика. - 2010.
4. Наследственная невропатия Шарко—Мари—Тута/ Шнайдер Н.А., Глущенко Е.В., Кантимирова Е.А. и др. - 2010.
Полиневропатии
Полиневропатии — гетерогенная группа заболеваний, характеризующаяся системным поражением периферических нервов. Полиневропатии подразделяются на первично аксональные и первично демиелинизирующие. Независимо от типа полинейропатии ее клиническая картина характеризуется развитием мышечной слабости и атрофии, снижением сухожильных рефлексов, различными нарушениями чувствительности (парестезиями, гипо- и гиперестезией), возникающими в дистальных отделах конечностей, вегетативными расстройствами. Важным диагностическим моментом при установлении диагноза полиневропатии является определение причины ее возникновения. Лечение полинейропатии носит симптоматический характер, основной задачей является устранение причинного фактора.
Полиневропатии — гетерогенная группа заболеваний, характеризующаяся системным поражением периферических нервов. Полиневропатии подразделяются на первично аксональные и первично демиелинизирующие. Независимо от типа полинейропатии ее клиническая картина характеризуется развитием мышечной слабости и атрофии, снижением сухожильных рефлексов, различными нарушениями чувствительности (парестезиями, гипо- и гиперестезией), возникающими в дистальных отделах конечностей, вегетативными расстройствами. Важным диагностическим моментом при установлении диагноза полиневропатии является определение причины ее возникновения. Лечение полинейропатии носит симптоматический характер, основной задачей является устранение причинного фактора или компенсация основного заболевания.
Причины полиневропатий
Независимо от этиологического фактора при полиневропатиях выявляют два типа патологических процессов — поражение аксона и демиелинизацию нервного волокна. При аксональном типе поражения возникает вторичная демиелинизация, при демиелинизирующем поражении вторично присоединяется аксональный компонент. Первично аксональными являются большинство токсических полиневропатий, аксональный тип СГБ, НМСН II типа. К первично демиелинизирующим полиневропатиям относятся классический вариант СГБ, ХВДП, парапротеинемические полиневропатии, НМСН I типа.
При аксональных полиневропатиях страдает главным образом транспортная функция осевого цилиндра, осуществляемая аксоплазматическим током, который несет в направлении от мотонейрона к мышце и обратно ряд биологических субстанций, необходимых для нормального функционирования нервных и мышечных клеток. В процесс вовлекаются в первую очередь нервы, содержащие наиболее длинные аксоны. Изменение трофической функции аксона и аксонального транспорта приводит к появлению денервационных изменений в мышце. Денервация мышечных волокон стимулирует развитие сначала терминального, а затем и коллатерального спраутинга, роста новых терминалей и реиннервацию мышечных волокон, что ведет к изменению структуры ДЕ.
При демиелинизации происходит нарушение сальтаторного проведения нервного импульса, в результате чего снижается скорость проведения по нерву. Демиелинизирующее поражение нерва клинически проявляется развитием мышечной слабости, ранним выпадением сухожильных рефлексов без развития мышечных атрофий. Наличие атрофий указывает на дополнительный аксональный компонент. Демиелинизация нервов может быть вызвана аутоиммунной агрессией с образованием антител к различным компонентам белка периферического миелина, генетическими нарушениями, воздействием экзотоксинов. Повреждение аксона нерва может быть обусловлено воздействием на нервы экзогенных или эндогенных токсинов, генетическими факторами.
На сегодняшний день общепринятой классификации полиневропатий не существует. По патогенетическому признаку полиневропатии разделяют на аксональные (первично поражение осевого цилиндра) и демиелинизирующие (патология миелина). По характеру клинической картины выделяют моторные, сенсорные и вегетативные полиневропатии. Однако в чистом виде эти формы наблюдаются весьма редко, чаще выявляют сочетанное поражение двух или трех видов нервных волокон (моторно-сенсорные, сенсорно-вегетативные др.).
По этиологическому фактору полиневропатии разделяют на наследственные (невральная амиотрофия Шарко-Мари-Тута, синдром Русси-Леви, синдром Дежерина-Сотта, болезнь Рефсума и пр.), аутоиммунные (синдром Миллера-Флешера, аксональный тип СГБ, парапротеинемические полиневропатии, паранеопластические невропатии и др.), метаболические (диабетическая полинейропатия, уремическая полиневропатия, печеночная полинейропатия и др.), алиментарные, токсические и инфекционно-токсические.
Симптомы полиневропатии
В клинической картине полиневропатии, как правило, сочетаются признаки поражения моторных, сенсорных и вегетативных волокон. В зависимости от степени вовлеченности волокон различного типа в неврологическом статусе могут преобладать моторные, сенсорные либо вегетативные симптомы. Поражение моторных волокон приводит к развитию вялых парезов, для большинства полиневропатий типично поражение верхних и нижних конечностей с дистальным распределением мышечной слабости, при продолжительных поражениях аксона развиваются мышечные атрофии. Для аксональных и наследственных полиневропатий характерно дистальное распределение мышечной слабости (чаще в нижних конечностях), которая более выражена в мышцах-разгибателях, чем в мышцах-сгибателях. При выраженной слабости перонеальной группы мышц развивается степпаж (т. н. «петушиная походка»).
Приобретенные демиелинизирующие полиневропатии могут проявляться проксимальной мышечной слабостью. При тяжелом течении может отмечаться поражение ЧН и дыхательных мышц, что чаще всего наблюдается при синдроме Гийена-Барре (СГБ). Для полиневропатий характерна относительная симметричность мышечной слабости и атрофии. Асимметричные симптомы характерны для множественных мононевропатий: мультифокальной моторной невропатии, мультифокальной сенсомоторной невропатии Самнера-Льюиса. Сухожильные и периостальные рефлексы при полиневропатии обычно снижаются или выпадают, в первую очередь снижаются рефлексы ахиллова сухожилия, при дальнейшем развитии процесса — коленные и карпорадиальные, сухожильные рефлексы с двуглавых и трехглавых мышц плеча могут оставаться сохранными длительное время.
Сенсорные нарушения при полиневропатии также чаще всего относительно симметричны, сначала возникают в дистальных отделах (по типу «перчаток» и «носков») и распространяются проксимально. В дебюте полиневропатии часто выявляют позитивные сенсорные симптомы (парестезия, дизестезия, гиперестезия), но при дальнейшем развитии процесса симптомы раздражения сменяются симптомами выпадения (гипестезия). Поражение толстых миелинизированных волокон приводит к нарушениям глубокомышечной и вибрационной чувствительности, поражение тонких миелинизированных волокон — к нарушению болевой и температурной чувствительности кожи.
Нарушение вегетативных функций наиболее ярко проявляется при аксональных полиневропатиях, так как вегетативные волокна являются немиелизированными. Чаще наблюдают симптомы выпадения: поражение симпатических волокон, идущих в составе периферических нервов, проявляется сухостью кожных покровов, нарушением регуляции сосудистого тонуса; поражение висцеральных вегетативных волокон приводит к дизавтономии (тахикардия, ортостатическая гипотензия, снижение эректильной функции, нарушение работы ЖКХ).
Диагностика полиневропатий
При выявлении медленно прогрессирующей сенсомоторной полиневропатии, дебютировавшей с перонеальной группы мышц, необходимо уточнить наследственный анамнез, особенно наличие у родственников утомляемости и слабости мышц ног, изменений походки, деформации стоп (высокий подъем). При развитии симметричной слабости разгибателей кисти необходимо исключить интоксикацию свинцом. Как правило, токсические полиневропатии характеризуются, помимо неврологических симптомов, общей слабостью, повышенной утомляемостью и редко абдоминальными жалобами. Кроме того, необходимо выяснить, какие препараты принимал/принимает пациент для того, чтобы исключить лекарственную полиневропатию.
Медленно прогрессирующее развитие асимметричной слабости мышц — клинический признак мультифокальной моторной полиневропатии. Для диабетической полиневропатии характерна медленно прогрессирующая гипестезия нижних конечностей в сочетании с чувством жжения и другими проявлениями в стопах. Уремическая полиневропатия возникает, как правило, на фоне хронического заболевания почек (ХПН). При развитии сенсорно-вегетативной полиневропатии, характеризующейся жжением, дизестезиями, на фоне резкого уменьшения массы тела необходимо исключить амилоидную полиневропатию.
Для наследственных полиневропатий характерны преобладание слабости разгибателей мышц стоп, степпаж, отсутствие ахилловых сухожильных рефлексов, высокий свод стопы. В более поздней стадии заболевания отсутствуют коленные и карпорадиальные сухожильные рефлексы, развиваются атрофии мышц стоп, голеней. Поражение мышц, соответствующее иннервации отдельных нервов, без сенсорных нарушений характерно для множественной моторной полиневропатии. В большинстве случаев преобладает поражение верхних конечностей.
Сенсорные полиневропатии характеризуются дистальным распределением гипестезий. В начальных стадиях заболевания возможна гиперестезия. Сенсомоторные аксональные невропатии характеризуются дистальными гипестезиями и дистальной мышечной слабостью. При вегетативных полиневропатиях возможны как явления выпадения, так и раздражение вегетативных нервных волокон. Для вибрационной полиневропатии типичны гипергидроз, нарушения сосудистого тонуса кистей, для диабетической полиневропатии, напротив, сухость кожных покровов, трофические нарушения, вегетативная дисфункция внутренних органов.
Исследование антител к GM1-гангликозидам рекомендуют проводить у пациентов с моторными невропатиями. Высокие титры (более 1:6400) специфичны для моторной мультифокальной невропатии. Низкие титры (1:400-1:800) возможны при хронической воспалительной демиелинизирующей полирадикулоневропатии (ХВДП), синдроме Гийена-Барре и иных аутоиммунных невропатиях. Следует помнить, что повышенный титр антител к GM1-гангкликозидам выявляют у 5% здоровых людей (особенно пожилого возраста). Антитела к ассоциированному с миелином гликопротеину выявляют у 50% пациентов с диагнозом «парапротеинемическая полиневропатия» и в некоторых случаях других аутоиммунных невропатий.
При подозрении на полиневропатии, связанные с интоксикацией свинцом, алюминием, ртутью проводят анализы крови и мочи на содержание тяжелых металлов. Возможно проведение молекулярно-генетического анализа на все основные формы НМСН I, IVA, IVB типов. Проведение игольчатой электромиографии при полиневропатиях позволяет выявить признаки текущего денервационно-реиннервационного процесса. Прежде всего, необходимо исследовать дистальные мышцы верхних и нижних конечностей, а при необходимости и проксимальные мышцы. Проведение биопсии нервов оправдано только при подозрении на амилоидную полиневропатию (выявление отложений амилоида).
Лечение полиневропатий
При наследственных полиневропатиях лечение носит симптоматический характер. При аутоиммунных полиневропатиях цель лечения заключается в достижении ремиссии. При диабетической, алкогольной, уремической и других хронических прогрессирующих полиневропатиях лечение сводится к уменьшению выраженности симптоматики и замедлению течения процесса. Один из важных аспектов немедикаментозного лечения — лечебная физкультура, направленная на поддержание мышечного тонуса и предупреждение контрактур. В случае развития дыхательных нарушений при дифтерийной полиневропатии может потребоваться проведение ИВЛ. Эффективного медикаментозного лечения наследственных полиневропатий не существует. В качестве поддерживающей терапии используют витаминные препараты и нейротрофические средства. Впрочем, эффективность их до конца не доказана.
Для лечения порфирийной полиневропатии назначают глюкозу, которая обычно вызывает улучшение состояния пациента, а также обезболивающие и другие симптоматические препараты. Медикаментозное лечение хронической воспалительной демиелинизирующей полиневропатии включает в себя проведение мембранного плазмафереза, применение иммуноглобулина человеческого или преднизолона. В ряде случаев эффективность плазмафереза и иммуноглобулина оказывается недостаточной, поэтому, если нет противопоказаний, лечение следует сразу начинать с глюкокортикостероидов. Улучшение наступает, как правило, через 25-30 дней; через два месяца можно начинать постепенное снижение дозы до поддерживающей. При снижении дозы глюкокортикостероидов необходимо проведение ЭМГ-контроля. Как правило, полностью отменить преднизолон удается в течение 10-12 месяцев, при необходимости можно «подстраховаться» азатиоприном (либо циклоспорин, либо микофенолата мофетил).
Лечение диабетической полиневропатии проводится совместно с эндокринологом, основной его целью является поддержание нормального уровня сахара крови. Для купирования болевого синдрома применяют трициклические антидепрессанты, а также прегабалин, габапентин, ламотриджин, карбамазепин. В большинстве случаев применяют препараты тиоктовой кислоты и витамины группы В. Регресс симптомов на ранней стадии уремической полиневропатии достигается нефрологами при коррекции уровня уремических токсинов в крови (программный гемодиализ, трансплантация почки). Из лекарственных средств применяются витамины группы В, при выраженном болевом синдроме — трициклические антидепрессанты, прегабалин.
Основной терапевтический подход в лечении токсической полиневропатии — прекращение контакта с токсическим веществом. При дозозависимых лекарственных полиневропатиях необходимо скорректировать дозу соответствующего лекарственного препарата. При подтвердившемся диагнозе «дифтерия» введение антитоксической сыворотки уменьшает вероятность развития дифтерийной полиневропатии. В редких случаях в связи с развитием контрактур и деформации стоп может понадобиться хирургическое лечение. Однако следует помнить, что длительная обездвиженность после оперативного вмешательства может негативно повлиять на двигательные функции.
Прогноз при полиневропатии
При хронической воспалительной демиелинизирующей полирадикулоневропатии прогноз на жизнь достаточно благоприятный. Летальность очень низкая, однако, полное выздоровление наступает очень редко. До 90% пациентов на фоне иммуносупрессивной терапии достигают полной либо неполной ремиссии. В то же время заболевание склонно к обострениям, применение иммуносупрессивной терапии может быть в виду ее побочных действий, приводящих к многочисленным осложнениям.
При наследственных полиневропатиях редко удается добиться улучшения состояния, так как заболевание медленно прогрессирует. Однако пациенты, как правило, адаптируются к своему состоянию и в большинстве случаев до самых поздних стадий заболевания сохраняют способность к самообслуживанию. При диабетической полиневропатии прогноз на жизнь благоприятный при условии своевременного лечения и тщательного контроля гликемии. Лишь в поздних стадиях заболевания выраженный болевой синдром способен значительно ухудшить качество жизни пациента.
Прогноз на жизнь при уремической полиневропатии полностью зависит от выраженности хронической почечной недостаточности. Своевременное проведение программного гемодиализа либо трансплантация почки способны привести к полному либо почти полному регрессу уремической полиневропатии.
Синдром Русси-Леви
Синдром Русси-Леви — наследственное заболевание из группы мотосенсорных невропатий, по некоторым представлениям являющееся фенотипическим вариантом амиотрофии Шарко-Мари-Тута. Основные симптомы включают сенситивную атаксию, слабость и гипотрофию мышц дистальных отделов конечностей (преимущественно нижних), сухожильную арефлексию. При установлении диагноза опираются на особенности клинического симптомокомплекса и течения заболевания, семейный анамнез, результаты электронейромиографии. Пациентам показано регулярное симптоматическое лечение: антихолинэстеразные фармпрепараты, витамины, калий, АТФ, массаж, физиопроцедуры, ЛФК.
Синдром Русси-Леви носит эпонимическое название в честь описавших его в 1926 году французских медиков невролога Леви и патолога Русси. Авторы обозначили описанный синдром как семейную арефлекторную дистазию с медленным прогрессированием. Базисными клиническими проявлениями синдрома выступают атаксия и сухожильная арефлексия, что легло в основу другого его названия - «синдром атаксии-арефлексии». Согласно международной классификации 10-го пересмотра, синдром Русси-Леви отнесен к группе наследственных мотосенсорных невропатий (НМСН). Однако среди клиницистов нет однозначного мнения, как классифицировать данную патологию. В соответствии с симптоматикой ряд специалистов в области неврологии относит синдром Русси-Леви к клинической форме, занимающей промежуточное положение между болезнью Шарко-Мари-Тута и атаксией Фридрейха, другие авторы считают его отдельным клиническим вариантом спинальной амиотрофии. Не смотря на полученные генетические доказательства последнего утверждения, многие неврологи до сих пор выделяют синдром Русси-Леви как отдельную нозологию.
Причины синдрома Русси-Леви
Семейный характер заболевания указывает на его наследственный генез. Ранее было установлено, что невропатия Русси-Леви встречается в семьях с повторными случаями болезни Шарко-Мари-Тута. Молекулярно-генетические исследования последних лет выявили, что генетическим субстратом синдрома выступают нарушения в 17-й хромосоме (локус 17р11.2) в виде наличия дубликатов отдельных генов, точковые мутации гена MPZ — нулевого миелинового протеина. Это открытие позволяет считать синдром Русси-Леви фенотипической формой невральной амиотрофии Шарко-Мари-Тута (НМСН вариант IА).
Наследование синдрома осуществляется аутосомно-доминантным путем. Морфологически определяются дегенеративные изменения задних спинальных корешков и периферических нервных стволов в виде умеренной демиелинизации и уменьшения числа нервных волокон; дистрофические изменения пирамидных и мозжечковых трактов, проходящих в боковых столбах спинного мозга; дегенерация задних столбов; атрофия мышечных волокон скелетной мускулатуры.
Симптомы синдрома Русси-Леви
Дебют симптоматики обычно приходится на детский возраст, иногда — пубертат, в отдельных случаях возможен у взрослых. Первоначальной жалобой выступает неловкость при ходьбе за счет снижения силы мышц голени (в большей степени перонеальной группы) и сенситивной атаксии. Мышечная слабость обуславливает затрудненное тыльное сгибание стоп, их свисание в состоянии подошвенного сгибания. Ранним признаком заболевания является выпадение сухожильных рефлексов нижних конечностей (коленных, ахилловых). Со временем наблюдается арефлексия верхних конечностей, слабость мышц дистальных отделов рук и атрофии мышц кисти. Отмечаются полиневритические нарушения чувствительности: болевая и температурная гипестезия в виде «носков» и «перчаток», расстройство глубоких видов (мышечно-суставного, вибрационного восприятия). Последнее обуславливает существование сенситивной атаксии.
Во внешнем виде пациента обращает на себя внимание гипотрофия мышц голеней, т. н. «ноги аиста». Выраженность нарушения походки вариабельна: от легкой неловкости до типичного степпажа с высоким подъемом согнутой в колене ноги и свисанием стопы. Атаксия редко достигает грубого выражения, характеризуется неустойчивостью при ходьбе, общей неловкостью, несоразмерностью и асимметричностью точных движений. Наблюдается шаткость в позе Ромберга, промахивание и тремор при выполнении координаторных проб. Характерна полая стопа, отличающаяся от стопы Фредрейха сочетанием с арахнодактилией, расположением пальцев в форме трезубца или двузубца, деформациями пальцев. Типично исчезновение высокого свода стопы при опоре на нее во время ходьбы. Наряду с деформацией стоп наблюдается похожая деформация кистей. У многих пациентов имеются деформации грудной клетки, искривление позвоночника по типу кифосколиоза и прочие маркеры соединительнотканной дисплазии (гипертелоризм, расширенная переносица, гидроцефальный череп, готическое небо).
Диагностика синдрома Русси-Леви
Диагноз базируется на клинической картине заболевания и ее изменении в динамике. От невральной амиотрофии невропатию Русси-Леви отличает меньшая выраженность мышечных гипотрофий, от болезни Фридрейха — отсутствие дизартрии и склонность к стабилизации состояния. Дифдиагностика проводится также с другими видами полиневропатий, с миопатией и миотонией, боковым амиотрофическим склерозом, болезнью Рефсума, миелопатиями. Пациентам показана консультация генетика и генеалогический анализ.
С целью уточнения диагноза невролог назначает электронейромиографию, результаты которой говорят о замедлении проведения импульсов по нервным стволам. В сложных диагностических случаях проводится биопсия мышц и/или поверхностных нервов нижних конечностей. Микроскопическое исследование мышечной ткани выявляет атрофические изменения отдельных пучков мышечных волокон. При микроскопии препаратов нерва определяется разрастание элементов шванновской оболочки, приводящее к гипертрофии нервных волокон, по своей форме напоминающей луковицу.
Лечение и прогноз синдрома Русси-Леви
В настоящее время разработана только симптоматическая терапия, базирующаяся на фармпрепаратах, облегчающих проведение нервного импульса и улучшающих обменные процессы нервной ткани. Составляющими лечения обычно выступают тиамин, пиридоксин, витамин С, цианокобаламин, оротат калия, антихолинэстеразные средства (амбенония хлорид, дистигмин, галантамин, пиридостигмин, неостигмин), АТФ. Параллельно рекомендованы занятия лечебной физкультурой, массаж, физиолечение и сегментарно-рефлекторная терапия. Деформация стоп является показанием к консультации ортопеда с подбором ортопедической обуви.
Синдром Русси-Леви отличается хорошим для жизни прогнозом. Медленное нарастание симптоматики с последующей стабилизацией процесса обеспечивает большинству пациентов длительную сохранность трудоспособности и негрубое ее снижение. В некоторых случаях наблюдается неуклонное прогрессирование мышечной слабости и гипотрофии, приводящее к затруднению самостоятельной ходьбы.
Читайте также:
- Симптомы и клиника болезней средней черепной ямки
- Лечение травматических стриктур прямой кишки. Операции при травматических стриктурах прямой кишки.
- Гемосидероз легких. Эхинококкоз легких. Доброкачественные опухоли легких.
- Синдром Хаммонда (Hammond)
- Инструктивная функция врожденного иммунитета. Контроль за формированием адаптивного иммунитета