Врожденная эритропоэтическая порфирия (болезнь Гюнтера) - клиника, диагностика, прогноз
Добавил пользователь Владимир З. Обновлено: 22.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Порфирия: причины появления, симптомы, диагностика и способы лечения.
Определение
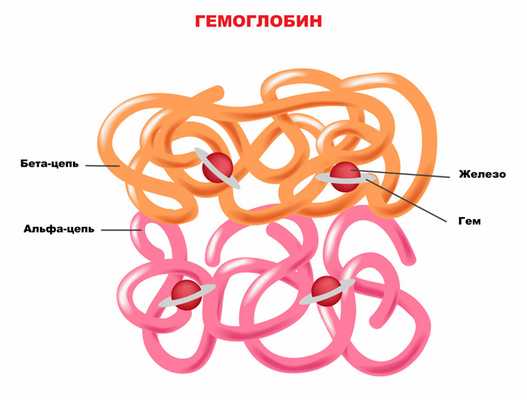
Порфирии (от греч. porphyreis - пурпурный) - это ряд заболеваний обмена веществ, при которых нарушается образование гема, представляющего собой комплексное соединение порфиринов с двухвалентным железом. В результате в организме происходит накопление порфиринов (пигментов, представляющих собой производные порфина) или их предшественников. К счастью, эти патологии встречаются относительно редко ‒ не более 7-12 случаев на 100 000 человек.
Причины появления порфирии
В подавляющем большинстве случаев причиной порфирий становятся генетические мутации, обусловливающие неполноценность активности того или иного фермента, участвующего в синтезе гема. Исключением является поздняя кожная порфирия (спорадическая форма), которая развивается на фоне заболеваний печени (алкогольного гепатита, вирусного гепатита С) или длительной интоксикации тяжелыми металлами.
Наследование порфирий происходит по аутосомно-доминантному (наследуется одна нормальная и одна измененная копия гена, причем измененная копия доминирует и «подавляет» нормальную, в результате чего развивается генетическое заболевание) или аутосомно-рецессивному типу (болезнь Гюнтера). Синтез гема проходит 8 последовательных этапов, за каждый из которых отвечает конкретный фермент, кодируемый определенным геном. Соответственно, для каждой формы порфирии существует специфичный ферментативный дефект.
Наибольшее количество гемов образуется в печени и костном мозге. В печени гемы входят в состав белков, участвующих в клеточном дыхании, расщеплении токсичных свободных радикалов и «обезвреживании» различных ксенобиотиков (чужеродных химических веществ, попадающих в организм извне). В костном мозге гемы необходимы для синтеза гемоглобина.
Результатом снижения активности ферментов становится торможение синтеза гемов, что ведет к накоплению его токсичных промежуточных метаболитов.
Провоцирующими факторами развития порфириновой болезни становятся:
- избыточная инсоляция;
- недостаточное питание и скудный рацион;
- систематические стрессы;
- чрезмерное употребление алкоголя;
- вирусные и бактериальные инфекции;
- хронические интоксикации солями тяжелых металлов.
- Беременность.
В отдельных случаях манифестация патологического процесса может произойти на фоне приема некоторых лекарственных препаратов: антибиотиков, антиконвульсантов, нестероидных противовоспалительных средств, пероральных контрацептивов.
Классификация заболевания
В основу разных классификаций порфирий положены различные критерии: клиническая симптоматика, локализация нарушения метаболизма порфиринов или тканевая тропность.
Современная классификация порфирий весьма разветвленная:
I. Эритропоэтические порфирии.
- Эритропоэтическая уропорфирия (врожденная эритропоэтическая порфирия, или болезнь Гюнтера).
- Эритропоэтическая протопорфирия:
- манифестная форма;
- латентная форма.
- Эритропоэтическая копропорфирия.
- Пирролопорфирия (острая перемежающая порфирия, доминантная порфирия шведского типа):
- манифестная форма;
- латентная форма.
- Протокопропорфирия (варигатная, или смешанная порфирия, доминантная порфирия Южно-Африканского типа):
- кожная форма;
- острая форма без кожных проявлений;
- комбинированная форма с кожными и острыми проявлениями;
- латентная форма.
- Урокопропорфирия (поздняя кожная порфирия):
- манифестная приобретенная (симптоматическая) форма (развивается при интоксикациях гексахлорбензолом, опухолях печени и других патологических состояниях;
- манифестная наследственная форма (развивается у лиц с наследственной предрасположенностью);
- латентная наследственная форма (выявляется у родственников больных).
- Наследственная копропорфирия.
- Неклассифицированная печеночная порфирия.
- Гепатоэритропоэтическая порфирия.
- Неклассифицированная печеночная порфирия, протекающая с клиническим синдромом световой оспы.
- Патологические состояния, сопровождающиеся геморрагическим синдромом и нарушениями порфиринового обмена печеночного типа.
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
- Эритропоэтические. К ним относятся врожденная эритропоэтическая порфирия (ВЭП, болезнь Гюнтера), эритропоэтическая протопорфирия (ЭПП). Основной клинический признак - поражение участков кожи, которые подвергаются воздействию прямых солнечных лучей (фотосенсибилизация). Данные патологии являются наиболее тяжелыми и имеют самый высокий процент летальности.
- Острые печеночные. Сюда включены острая перемежающаяся порфирия (ОПП), вариегатная порфирия (ВП), наследственная копропорфирия (НКП). Острые порфирии характеризуются приступообразным течением. Преимущественно страдает нервная система. При ВП и НКП также встречаются признаки фотосенсибилизации.
- Хронические печеночные. К ним относят позднюю кожную порфирию (ПКП), которая имеет наследственную и приобретенную формы. Это наиболее благоприятный вид порфирии.
- Клинико-биохимические анализы. При болезни Гюнтера в общем и биохимическом анализах крови выявляются признаки гемолиза (пойкилоцитоз, анизоцитоз, сфероцитоз, ретикулоцитоз, повышение непрямого билирубина и сывороточного железа), увеличение печеночных трансаминаз. У больных с острыми порфириями отмечается снижение уровня глюкозы и натрия, при ПКП - увеличение сывороточного железа и ферритина. Также у 80% с ПКП выявляются положительные маркеры вируса гепатита С.
- Специфические исследования. Для диагностики острых порфирий широко используется скрининговая проба Эрлиха (при смешивании специального реактива с мочой она окрашивается в красный цвет). Для ОПП характерно повышение ДАЛК и порфобилиногена в моче, для ВП - протопорфирина в кале, для НКП - копропорфирина в кале и моче. При ПКП в моче увеличено содержание уропорфирина, в кале - копропорфирина. При ЭПП наблюдается высокая концентрация протопорфирина в эритроцитах и кале, при ВЭП - уропорфирина в моче, кале и эритроцитах. При люминесцентной микроскопии плазма дает красное флуоресцирование при ПКП и эритропоэтических порфириях.
- Острые. Для подавления образования порфобилиногена и ДАЛК применяют гем-аргинат, производные АТФ (аденил, рибоксин) и большие дозы глюкозы с дальнейшим переходом на высокоуглеводную диету. Для купирования вегетативной симптоматики используют октреотид, для ускорения восстановления миелина в нервных волокнах - витамины группы В. При менструалозависимых атаках эффективна овариосупрессивная терапия. С этой целью назначают агонисты гонадотропин-рилизинг гормона.
- Эритропоэтические. Данные порфирии очень плохо поддаются терапии. Основное лечение - это защита кожных покровов от солнечного света (окна со стеклом, не пропускающим ультрафиолет, закрытая одежда, фотозащитные кремы, прием бета-каротина). Необходимо обрабатывать эрозии антисептическими растворами для профилактики инфекционных осложнений. При выраженном гемолизе показана спленэктомия. В некоторых случаях болезни Гюнтера эффективной оказывается трансплантация костного мозга. При ЭПП дополнительно назначаются гепатопротекторы (урсодезоксихолевая кислота, адеметионин) и антицирротическая терапия.
- ПКП. С целью удаления порфиринов и избытка железа проводят плазмаферез и флеботомию (кровопускания). При противопоказаниях к данным процедурам назначаются аминохинолиновые (хлорохин) и комплексообразующие (дефероксамин) препараты. Для уменьшения всасывания порфиринов в желудочно-кишечном тракте применяют энтеросорбенты (активированный уголь). Также используются солнцезащитные кремы. При наличии гепатита С необходима противовирусная терапия интерфероном-альфа и рибавирином.
- по месту нарушения метаболизма порфиринов различают: эритропоэтические (первичное нарушение в костном мозге) и печеночные порфирии (нарушения развиваются первично в печени);
- по клиническим проявлениям порфирии делятся на острые формы, проявляющиеся преимущественным поражением нервной системы и формы, манифестирующие поражением кожи.
- Повышенная чувствительность к солнечной радиации, свету с появлением на открытых участках кожи пузырей.
- Отсутствие типичной симптоматики для острой порфирии (абдоминальных симптомов).
- Чрезмерное развитие волосяного покрова.
- Выделение окрашенной в красный цвет мочи.
- Розовато-коричневая окраска зубов.
- Увеличение селезенки на фоне гемолитической анемии.
- Болевой синдром — интенсивные боли в животе, сопровождаемые тошнотой/рвотой, расстройствами стула (диарея/запоры); боли носят приступообразный характер, реже - постоянный и могут продолжаться от 5-6 часов до нескольких дней с локализацией в различных отделах живота.
- Неврологическая симптоматика манифестирует вялыми параличами; парезами; полиневритом; сенсорными/бульбарными нарушениями, расстройством функции уретрального сфинктера.
- Психические расстройства (эмоциональная лабильность, хроническая бессонница, депрессии/тревоги, психомоторное возбуждение, бред, слуховые/зрительные галлюцинации, делирий).
- Гипоталамическая дисфункция (лихорадка центрального генеза, гипонатриемии).
- Эпилептиформные припадки с высоким риском развития во время острых приступов коматозного состояния.
- Нарушения со стороны сердечно-сосудистой системы — гипертензия, синусовая тахикардия, изменения на ЭКГ.
- Пигментурия (розовая/красная моча, темная диффузная окраска кожи, хлоазмы, веснушки).
- Для латентной формы характерны мышечная слабость, периодически возникающие боли в животе, синусовая тахикардия, бессонница, гипертензия, реже — психологические изменения личности.
- Манифестные острая форма перемежающейся порфирии может протекать в нескольких вариантах.
Лёгкая форма характеризуется непродолжительными периодически возникающими острыми приступами болезни, ограничивающиеся абдоминальными симптомами, которые заканчиваются в большинстве случаев благоприятно.
Тяжёлая форма: характерны тяжёлые приступы длительностью от 2 до 10 недель с рецидивами через несколько месяцев/1-2 года. Манифестирует абдоминальными симптомами, психическими расстройствами/неврологическими нарушениями. Может закончиться летально.
Ступенчатая форма: для нее характерным является нарастание симптоматики и глубоких общих нарушений с каждым новым приступом. Рецидивы учащаются и наступают через 1-2 месяца. В большинстве случаев исход неблагоприятный и каждый 3-4 приступ заканчивается летально.
Острейшая форма: крайне тяжелое течение, сопровождаемое тяжелыми общими нарушениями; чаще встречается во время беременности и 24-60% случаев заканчивается летально от паралича дыхательного центра.
Симптомы порфирии
Сроки дебюта рассмотренных выше форм заболевания различны: эритропический тип проявляется в возрасте 3-5 лет, острый печеночный — в 14-16 лет, хронический печеночный — после 40 лет. После манифестации патологии пациенты сталкиваются со специфической симптоматикой.
При острых формах порфирии больные жалуются на сильные боли в животе, задержку стула, учащенное сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого).
Тяжесть состояния пациента в основном обусловлена неврологическими симптомами - болью и снижением чувствительности по всему телу, прогрессирующей мышечной слабостью, судорожными припадками, различными психическими расстройствами (тревожностью, психомоторным возбуждением, бредом).
При поздней кожной форме на участках кожного покрова, подвергающихся постоянному воздействию солнечного света, формируется гиперпигментация, в результате чего кожа приобретает землистый или бронзовый оттенок.
Выраженными симптомами этой формы порфирии являются везикулезные и буллезные высыпания, которые покрыты корками, шелушение кожи, медленно заживающие эрозии, милиумы.
Кроме того, могут отмечаться явления гипертрихоза (избыточного оволосения) лобно-височной области лица, фотосенсибилизация.
При эритропоэтических порфириях наблюдаются признаки светочувствительности, причем даже более отчетливые, чем при поздней кожной порфирии. Пациент испытывает сильную боль, если на кожный покров попадают прямые солнечные лучи. Обширные эрозии оставляют после себя атрофичексие рубцы, что приводит к обезображиванию внешнего вида больного. В результате множественных атрофических рубцов на коже кистей рук развиваются контрактуры. Избыточное количество порфиринов приводит к тому, что зубная эмаль приобретает красновато-коричневый цвет (эритродонтия), моча становится красной или розовой.
Специфический признак эритропоэтической порфирии - утолщение, огрубение и уплотнение кожи периоральной и периорбитальной зон, крыльев и спинки носа, тыльной поверхности кистей.
Диагностика порфирии
При постановке диагноза учитывается наличие данного заболевания у близких родственников, возраст больного, обстоятельства возникновения симптомов (инсоляция, прием лекарств или алкоголя, голодание, инфекции, беременность).
- Общеклинический № и биохимический анализ крови: АЛТ, АСТ, непрямой билирубин, сывороточное железо, ферритин.
Синонимы: Общий анализ крови, ОАК. Full blood count, FBC, Complete blood count (CBC) with differential white blood cell count (CBC with diff), Hemogram. Краткое описание исследования Клинический анализ крови: общий.
Диагностика и лечение острых порфирий
НАЦИОНАЛЬНЫЕ КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ
ДИАГНОСТИКА И ЛЕЧЕНИЕ ОСТРЫХ ПОРФИРИЙ
Москва 2018 г.
Порфирии представляют собой группу заболеваний, состоящую из семи нозологических форм. Причинами их возникновения являются генетически обусловленные нарушения активности различных ферментов в цепи биосинтеза гема (Рисунок 1, Приложения). Это приводит к нарушению обмена порфиринов, имеющему свои особенности при каждой форме порфирии и определяющему их клинические проявления. Начало изучения нарушений порфиринового обмена относится к 1841г., когда Scherer доказал, что красный цвет мочи больных обусловлен наличием в ней определѐнных пигментов, а не присутствием молекул гемоглобина. Fisher в 1930г. получил Нобелевскую премию за работу по изучению промежуточных продуктов гема и в 1934 году опубликовал книгу «Химия пирролов» [6]. Схема биосинтеза гема была описана в 50-е годы прошлого столетия.
В дальнейшем были идентифицированы все восемь ферментов в этом цикле [5]. Установлено, что каждая нозологическая форма порфирий связана с дефектом активности одного из ферментов (кроме синтетазы дельта-аминолевулиновой кислоты (-АЛК)), закодированного в одном гене.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
Порфирии подразделяются на эритропоэтические и печѐночные в зависимости от ткани, где происходит преимущественное нарушение метаболизма порфиринов (см. классификация I) [10]. Вместе с тем, порфирии могут подразделяться на формы с поражением кожных покровов и острые, провоцируемые формы (см. классификация II).
- Поздняя кожная порфирия
Острые формы порфирий характеризуются яркой неврологической и вегетативно-сосудистой симптоматикой, в основе которой лежит полинейропатия. При порфирии, обусловленной дефицитом дегидратазы δ-аминолевулиновой кислоты и острой перемежающейся порфирии (ОПП) нет кожных проявлений. Это объясняется тем, что у них активными метаболитами являются предшественники порфиринов (δ-АЛК и порфобилиноген (ПБГ)), которые не имеют сродства к тканям дермы.
При снижении активности ферментов поздних этапов биосинтеза гема происходит накопление собственно порфиринов, избыток которых в дерме приводит к фототоксическим реакциям. Вследствие этого клиника кожных поражений характерна для таких острых порфирий как: наследственная копропорфирия (НКП) и вариегатная порфирия (ВП), а также поздней кожной порфирии (ПКП) и эритропоэтических порфирий.
Этиология и патогенез
Развитие различных форм порфирий связано с нарушениями цикла биосинтеза гема и имеет общие черты. В основе развития каждой формы порфирии лежит генетически обусловленное снижение или отсутствие активности определѐнного фермента в цепи биосинтеза гема (рисунок). Гены ферментов расположены на разных хромосомах и не имеют групповой сцепленности. Снижение активности фермента до 50% от нормы может не иметь клинических проявлений.
При ОП реализовать генетическое носительство и спровоцировать клиническую манифестацию заболевания могут:
-лекарственные препараты (НПВС, барбитураты, цефалоспорины сульфаниламиды и др. список которых представлен в приложении)
Перечисленные факторы приводят к повышенному потреблению конечного продукта цикла биосинтеза - гема (например, активация системы цитохрома Р-450) [10], либо оказывают непосредственное стимулирующее воздействие на активность первого фермента цикла биосинтеза - синтетазы -АЛК, что приводит к повышению еѐ активности (например действие прогестерона) [7], в результате чего ускоряется синтез всех промежуточных продуктов метаболизма порфиринов. На этапе участия дефектного фермента начинается избыточное накопление метаболитов в токсических концентрациях, что приводит к обострению заболевания. При ОП избыточное накопление -АЛК и ПБГ в тканях приводит к сегментарной демиелинизации нервных волокон с нарушением нервной проводимости. Токсическому воздействию подвержены все отделы нервной системы человека.
Периферическая сенсорно-моторная полинейропатия является следствием вторичной демиелинизации нервных волокон.
Вовлечение вегетативной нервной системы имеет следующий патогенез: - поражение абдоминальных вегетативных сплетений сопровождается спазмом сосудов брыжейки и нарушением моторики кишечника.
- ослабление активности n. vagus приводит к преимущественному влиянию на сердечно-сосудистую систему симпатического отдела; этому также способствует 10-ти кратное увеличением экскреции катехоламинов и нарушение функции барорецепторов артериальных сосудов.
Нарушение функции центральной нервной системы является следствием токсического воздействия предшественников порфиринов на нейроны головного мозга и развития длительного спазма артериол, гипонатриемии и гипергидратации, что приводит к тяжѐлым энцефалопатиям.
Поздняя кожная порфирия (ПКП) и эритропоэтические порфирии имеют хроническое течение с периодами обострений, которые могут вызвать:
Повышенная светочувствительность кожных покровов связана с фотохимическими реакциями, спровоцированными порфиринами. Избыток порфиринов в коже подвергается активному воздействию спектра солнечного излучения с длинами волн 400 - 410 нм, что приводит к образованию реактивных частиц, например, супероксид аниона, активирующего ксантин-оксидазу, и других метаболитов, повреждающих клетки базальной мембраны. Повторные атаки приводят к развитию нескольких слоѐв базальных мембран и образованию пласта кровеносных сосудов в поверхностных слоях дермы. Реактивные кислородсодержащие частицы также могут приводить к высвобождению гистамина из тучных клеток, усиливая явления фототоксичности.
Изменения кожи. Уропорфириноген - основной метаболит при поздней кожной порфирии стимулирует синтез фибробластами коллагена. При эритропоэтической протопорфирии жирорастворимый протопорфириноген откладывается в стенке сосудов дермы, приводя к их утолщению.
Пигментирование кожи и гипертрихоз наблюдаются в периорбитальных областях при поздней кожной порфирии и эритропоэтических порфириях, однако, механизм этих изменений до конца не изучен.
Эпидемиология
Порфирии не являются эндемичными заболеваниями и с одинаковой частотой встречаются среди населения всех континентов. Частота встречаемости острых форм порфирий (ОП) по различным оценкам составляет 7-12 случаев на 100 тысяч здоровых людей. В то же время частота бессимптомного носительства генетических дефектов, приводящих к ОП, составляет ~ 50-100 случаев на 100000 человек.
Клиническая картина
Cимптомы, течение
-эритема, волдыри на открытых участках кожи.
Первый приступ острых порфирий может развиться в возрасте старше 14-16 лет, значительно чаще у женщин. Начало заболевания острое, реже подострое. После воздействия порфириногенных факторов появляются боли в животе, конечностях, пояснице, тошнота, рвота. К концу второй недели заболевания появляется мышечная слабость, переходящая в парезы и параличи. Характерны тахикардия (до 110-130 артериальная гипертензия (до 180/100), выделение мочи с красноватым оттенком, неадекватное поведение и галлюцинации. При отсутствии лечения в течение 20-40 дней у больных могут развиться бульбарные нарушения и паралич дыхательной мускулатуры. В анализах крови нередко выявляется гипонатриемия.
Диагностика
Лабораторная диагностика острых порфирий, кроме порфирии обусловленной дефицитом дегидратазы --АЛК основывается на определении в моче избытка порфобилиногена с помощью:
- качественного скрининг-теста свежего образца мочи больного с использованием реактива Эрлиха по методу Watson-Schwartz. При наличии в моче избытка ПБГ образуется окрашенный продукт розово-красного цвета;
- количественного определения содержания ПБГ в моче (норма не превышает 2 мг/л).
При порфирии, обусловленной дефицитом дегидратазы-АЛК в моче, определяется высокая концентрация дельта-аминолевулиновой кислоты при нормальном содержании ПБГ.
Диагноз острой порфирии устанавливается на основании характерной клинической картины и высокого содержания ПБГ или -АЛК в моче [1,17].
Дифференциальный диагноз между ОПП и ВП или НКП основывается при измерении содержания общих порфиринов в кале [1,17]. В норме концентрация порфиринов в кале менее 200 нмоль/г сухого веса. Нормальная концентрация общих порфиринов в кале подтверждает диагноз ОПП. Повышение концентрации в несколько раз свидетельствует в пользу НКП или ВП. При исследовании плазмы спектрофлюориметрическим методом можно дифференцировать НКП от ВП.
Следующим этапом диагностики является определение активности ферментов в клетках крови. Это позволяет подтвердить диагноз у больных с клиническими проявлениями заболеваний и выявить бессимптомных носителей. Несмотря на распространѐнность и удобство диагностики бессимптомного носительства порфирий путѐм оценки активности специфического фермента в клетках, этот метод не является абсолютно достоверным [8,6].
Поэтому заключительным этапом диагностики порфирий у больных и, в особенности, у бессимптомных носителей является проведение ДНК- анализа [2].
Причиной поражения кожных покровов может быть повышенная светочувствительность, как следствие одной из форм порфирий. Тем не менее, следует провести дифференциальную диагностику кожной порфирии с другими дерматологическими заболеваниями.
- определение общих порфиринов в плазме и исследование спектра ее поглощения при флюоресцентной спектроскопии;
Для подтверждения диагноза ЭПП следует провести дополнительный тест - определение общих порфиринов в эритроцитах. При повышенной их концентрации следует измерить соотношение свободного и Zn-связанного протопорфиринов. В случае значительного превышения концентрации свободного протопорфирина подтверждается диагноз ЭПП.
Если результаты всех перечисленных тестов у пациента с активными кожными нарушениями будут нормальными, то диагноз порфирии будет маловероятным..
В случае же получения хотя бы одного или нескольких положительных результатов подтверждается диагноз кожной порфирии, форма которой определяется с помощью диагностических тестов.
Дифференциальный диагноз
- соматизация и синдром хронической усталости
Первичный этап диагностики, на практике, является самым сложным и ответственным для врача. Вариативность течения и симптоматики ОП (особенно при атипичном и моносимптомном вариантах развития) создают значительные трудности в своевременной постановке этого диагноза, нередко уводя врача в сторону ошибочных предположений. Выполнение же скрининг теста на наличие избытка ПБГ в моче всем обращающимся без анализа первичных жалоб нерационально с точки зрения временных и материальных затрат. Опытным путѐм, подтверждѐнным статистическими данными, выделены характерные для ранних сроков течения и специфичные для ОП симптомы. Разработана шкала, содержащая данные анамнеза и симптомы как характерные для клинического течения острых порфирий, так и наиболее часто ложно приписываемых ОП. Скрининг имеющихся у пациентов симптомов и сопоставление этих симптомов с симптоматикой, представленной в таблице (приложение 1)*, позволяет по сумме баллов судить о степени вероятности наличия порфирии у больного. Такой подход позволяет минимизировать число ятрогений, связанных с назначением пациентам с неустановленным диагнозом порфириногенных препаратов.
*В шкале представлены симптомы, встречающиеся при острых порфириях, которым присвоено определенное количество баллов, имеющих как положительные, так и отрицательные значения в зависимости от частоты их встречаемости при острой порфирии. Эти баллы суммируются, и если сумма баллов составляет менее 5 - диагноз острой порфирии маловероятен, если от 5 до 15 баллов - диагноз порфирии возможен, если более 15 баллов, то диагноз острой порфирии высоко вероятен.
Поражение кожных покровов при порфириях следует дифференцировать с прочими дерматологическими заболеваниями.
Лечение
1. Острые порфирии, цель лечения - предупреждение атак заболевания и развития необратимых изменений нервной системы.
Профилактика развития приступов ОП предусматривает ограничение воздействия на организм провоцирующих факторов, а именно:
- приѐма лекарственных препаратов с повышенной порфириногенной активностью, инсоляции развития бактериальных и вирусных инфекции (особенно HCV, HBV, CMV), состояния гипогликемии
- у женщин с доказанной связью между наступлением menses и частыми приступами - предупреждение менструаций.
Если менструальные циклы часто (три и более раз в год) провоцируют атаки ОП, репродуктивную функцию необходимо подавлять, для чего используют оральные контрацептивы - Ригевидон, Овидон; ГнРГ- Золадекс, синарел; андрогены - сустанон, андриол; овариоэктомия в отдельных случаях. Отмена овариосупрессии производится после достижения длительного бесприступного (несколько месяцев и более) течения заболевания.
Лечение начинают при наличии нескольких симптомов ОП (см. выше) и повышении показателей порфиринового обмена (по сравнению с предыдущими данными в динамике). При развитии острого приступа необходимо немедленное начало патогенетической терапии для подавления избыточного биосинтеза порфиринов:
2. Обеспечение избыточного поступления в орагнизм углеводов (200-600гр сухого вещ-ва глюкозы). 40%-1000мл в/в капельно в сутки, ежедневно, 2-4 недели [4].
3. Сандостатин в дозе 100-500 мкг/сут., подкожно, ежедневно на протяжении от 4-х недель до 6 месяцев в сочетании с плазмаферезами 6-10 сеансов [13].
4. Рибоксин 2%-10мл в разведении на 100-200 мл 0.9% раствора NaCl, в/в капельно 1-2 раза в сутки, ежедневно 2-4 недели.
Порфирия ( Порфириновая болезнь )
Порфирии ‒ большая группа наследственных заболеваний, характеризующихся нарушением биосинтеза гема и накоплением его токсичных метаболитов. Клинические проявления крайне разнообразны - от светочувствительности и кожных высыпаний до болей в животе, полного паралича и острых психозов. Диагностика осуществляется с помощью молекулярно-генетических тестов, специальных лабораторных методов определения порфиринов и их предшественников в моче и кале, оценки активности ферментов в крови. Лечение заключается в мероприятиях, направленных на снижение образования токсических метаболитов, их выведение из крови, проведении симптоматической терапии и хирургических вмешательств.
МКБ-10
Общие сведения
Порфирии (от греч. «porphyreis» - пурпурный) - ряд заболеваний обмена веществ, при которых нарушается образование гема, в результате чего в организме накапливаются порфирины или их токсичные предшественники. Патологии данной группы встречаются относительно редко ‒ от 7 до 12 случаев на 100 000 человек. Отдельные нозологии имеют свою эндемичность. Так, распространенность поздней кожной порфирии в странах Южной Африки составляет 1:800, острой перемежающейся порфирии в Швеции ‒ 1:1000, вариегатной порфирии в Южной Африке ‒ 1:3000. У большинства порфирий нет гендерных различий, кроме поздней кожной формы (чаще страдают мужчины) и острой интермиттирующей (чаще болеют женщины).
Причины порфирий
В подавляющем большинстве случаев причиной порфирий выступают генетические мутации, обусловливающие неполноценность активности того или иного фермента, участвующего в биосинтезе гема. Исключением является поздняя кожная порфирия (спорадическая форма), которая развивается вследствие заболеваний печени (алкогольный гепатит, вирусный гепатит С) или длительной интоксикации тяжелыми металлами. Наследование порфирий происходит по аутосомно-доминантному или аутосомно-рецессивному типу. Синтезирование гема протекает в 8 последовательных этапов, за каждый отвечает свой фермент, кодируемый определенным геном. Для каждой формы порфирии существует специфичный ферментативный дефект.
Гем представляет собой комплексное соединение порфиринов с двухвалентным железом. Наибольшее количество гема образуется в печени и костном мозге. В печени гем входит в состав белков, участвующих в клеточном дыхании, расщеплении токсичных свободных радикалов и обезвреживании различных ксенобиотиков. В костном мозге гем используется для образования гемоглобина. Результатом сниженной активности ферментов является торможение синтеза гема на определенном уровне, что ведет к накоплению его токсичных промежуточных метаболитов.
Помимо генетической мутации, для развития острых порфирий необходимо воздействие провоцирующих факторов, стимулирующих выработку порфиринов. Такими факторами являются голодание, длительная инсоляция, стрессы, алкоголь, инфекции, интоксикация тяжелыми металлами (ртуть, свинец), лекарственные средства, подвергающиеся метаболизму системой цитохрома P-450 (нестероидные противовоспалительные препараты, антибиотики, антиконвульсанты, оральные контрацептивы, седативные средства). Особую роль играют колебания женских половых гормонов во время менструаций или беременности. У женщин месячные являются наиболее частым провоцирующим фактором, а беременность ассоциируется с тяжелым течением заболевания.
Патогенез
В результате неполноценности ферментов, участвующих в образовании гема, и действия провоцирующих факторов происходит увеличение концентрации его токсичных продуктов обмена. Для хронических порфирий характерно накопление протопорфирина, копропорфирина и упопорфирина. При острых формах возрастает количество порфобилиногена и дельта-аминолевулиновой кислоты (ДАЛК).
Порфирины накапливаются в коже и под действием ультрафиолетового излучения (солнечного света) запускают процесс перекисного окисления липидов, вызывая деструкцию и гибель клеток кожи. Копропорфирин и протопорфирин усиливают пигментацию кожи и ускоряют рост волос (гипертрихоз). Плохо растворимый в воде протопорфирин откладывается в клетках печени, закупоривает портальные тракты и желчные протоки. Отложение уропорфирина в эритроцитах приводит к их ускоренному разрушению в селезенке (гемолиз). Предшественники порфиринов (ДАЛК и порфобилиноген), накапливаясь в нервной ткани, вызывают демиелинизацию и аксональную дегенерацию нервных волокон.
В основу разных классификаций порфирий положены различные критерии: клиническая симптоматика, локализация нарушения метаболизма порфиринов или тканевая тропность. Наиболее целесообразно выделять следующие виды порфирий:
Симптомы порфирий
Спектр клинических проявлений очень широк. Порфирии могут протекать в виде острых атак или хронически. Различия наблюдаются также в возрасте дебюта заболевания. Так, эритропоэтические порфирии манифестируют уже в дошкольном детстве (3-5 лет), острые порфирии - после полового созревания (14-16 лет), а спорадическая (приобретенная) форма ПКП - после 40 лет.
При острых порфириях развиваются сильные боли в животе, задержка стула, учащение сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого). Тяжесть состояния пациента в основном обусловлена неврологическими симптомами - болью по всему телу, снижением чувствительности, прогрессирующей мышечной слабостью, иногда достигающей полного паралича, судорожными припадками, различными психическими расстройствами (тревожность, психомоторное возбуждение, бред, галлюцинации).
При поздней кожной форме возникает гиперпигментация участков кожи, подвергающихся постоянному воздействию солнечного света (лицо, шея, ушные раковины, верхняя часть груди, кисти рук). Кожа приобретает землистый или бронзовый оттенок. Также характерны гипертрихоз лобно-височной области лица, фотосенсибилизация, проявляющаяся повышенной ранимостью кожи и образованием пузырей с жидкостным содержимым. После вскрытия пузырей формируются эрозии. На местах разрешения эрозий образуются атрофические рубцы.
При эритропоэтических порфириях наблюдаются более выраженные признаки светочувствительности, чем при ПКП (ранимость, пузыри, эрозии). При длительном нахождении на свету появляется покраснение и сильное жжение кожи. Обширные эрозии оставляют после себя грубые рубцы на лице, что приводит к обезображиванию внешнего вида больного. В результате множественных рубцов на коже кистей рук развиваются контрактуры суставов, что значительно затрудняет их движения. Моча становится красной или розовой, а зубы окрашиваются в красно-коричневый цвет (эритродонтия). Из-за увеличенной селезенки могут появиться тяжесть или ноющие боли в левом подреберье. Специфический признак ЭПП - утолщение, огрубение и уплотнение кожи вокруг рта и глаз, на крыльях и спинке носа, на тыльных поверхностях кистей.
Осложнения
Нарушения порфиринового обмена ухудшают течение сердечно-сосудистых заболеваний, неблагоприятно влияют на углеводный метаболизм и повышают риск развития сахарного диабета 2 типа. Острые формы порфирий вследствие выраженной полинейропатии осложняются параличом дыхательной мускулатуры, аспирационной пневмонией, отеком головного мозга, тромбоэмболиями, рабдомиолизом. Постоянные эрозии кожных покровов могут привести к бактериальным инфекциям. При ЭПП из-за отложения нерастворимого в воде протопорфирина может развиться цирроз печени и печеночная недостаточность.
При подозрении на порфирию пациента направляют к врачу-гематологу. При постановке диагноза учитывается наличие заболевания у близких родственников, возраст больного, обстоятельства возникновения симптомов (инсоляция, прием лекарств или алкоголя, голодание, инфекции, менструации, беременность). Лабораторная диагностика порфирий следующая:
Также для подтверждения диагноза проводится определение уровня ферментов цикла биосинтеза гема в эритроцитах, лимфоцитах или плазме - порфобилиногендезаминазы (ОПП), копропорфириноген-оксидазы (НКП), протопорфириноген-оксидазы (ВП), уропорфириногенсинтетазы (ВЭП), уропорфириногендекарбоксилазы (ПКП), феррохелатазы (ЭПП). Заключительным этапом диагностики является молекулярно-генетическое тестирование для выявления мутаций генов, кодирующих перечисленные выше ферменты. Данные исследования особенно эффективны для распознавания асимптомных форм порфирий.
Эритропоэтические порфирии дифференцируют с дерматологическими заболеваниями (буллезным пемфигоидом, вульгарной пузырчаткой), с гематологическими патологиями, протекающими со спленомегалией (лейкозами, лимфомами, аутоиммунными гемолитическими анемиями) с болезнями почек. ПКП дифференцируют с заболеваниями печени, гемохроматозом, надпочечниковой недостаточностью. Острые порфирии следует дифференцировать с хирургическими заболеваниями, сопровождающимися сильной болью в животе, неврологическими и психиатрическими патологиями.
Лечение порфирий
Пациентов с острыми и эритропоэтическими порфириями необходимо госпитализировать отделение гематологии. Лечение ПКП возможно как в стационаре, так и в амбулаторных условиях. На сегодняшний день не существует эффективных методов, полностью ликвидирующих нарушения обмена порфиринов. Основной упор делается на патогенетическую и симптоматическую терапию, а также на устранение провоцирующих факторов. Способы лечения зависят от вида порфирий:
Прогноз и профилактика
В большинстве случаев порфирии являются тяжелыми заболеваниями с неблагоприятным прогнозом. При эритропоэтических формах продолжительность жизни составляет около 30 лет, смерть наступает от интеркуррентных инфекций. ЭПП часто приводит к циррозу печени. При атаках острых порфирий летальный исход наблюдается в 15-20%, основная причина смерти - паралич дыхательных мышц. При ПКП прогноз благоприятный, тяжелых осложнений не происходит. Для предупреждения рецидивов рекомендуется избегать провоцирующих факторов - инфекций, голодания, стрессов, длительной инсоляции, употребления алкоголя и определенных лекарственных средств. Людям, у которых в семье есть больной порфирией, необходимо определять активность ферментов цикла синтеза гема и проводить ДНК-диагностику для выявления генетических мутаций.
1. Заболевания внутренних органов при манифестных и латентных нарушениях порфиринового обмена/ Кривошеев Б.Н. и др. - 2014.
2. Диагностика и лечение острых порфирий: Клинические рекомендации национального гематологического сообществ/ под ред. Пустовойт Я.С., Кравченко С.К., Шмакова Р.Г., Савченко В.Г. - 2018.
3. Диагностическая роль отдельных синдромов и симптомов в семиотике острых порфирий/ Пустовойт Я.С., Галстян Г.М., Савченко В.Г.//Гематология и трансфузиология. - 2014 - №59(3).
Порфирия
Болезнь Гюнтера (порфирия)

Болезнь порфирия (син. порфириновая болезнь) представляет собой большую группу заболеваний обмена веществ, обусловленных преимущественно наследственным дефектом в системе биосинтеза гема (соединение ионов железа с производными порфирина) и накоплением в организме его токсичных метаболитов (порфобилиногена/δ-аминолевулиновой кислоты). Большинство порфирий являются врождёнными заболеваниями с наследованием по аутосомно-доминантному типу. Значительно реже порфирии, обусловленные нарушением метаболизма, являются приобретенным и развиваются под воздействием различных факторов, способствующих процессу ингибирования ферментов синтеза гема.
В человеческом организме происходит постоянный эндогенный синтез пуринов/пиримидинов. К пуринам относятся ксантин, аденин, гипоксантин, гуанин; к пиримидинам - цитозин, урацил, тимин, оротовая кислота. Они необходимы для хранения, транскрипции/трансляции генетической информации, деления/роста клеток, передачи сигналов и накопления энергии.
Порфирины в человеческом организме синтезируются в клетках костного мозга, печени, тканях нервной системы, поджелудочной железе и находятся как в свободном, так и связанном состоянии, образуя различные сложные белковые соединения с ионами железа (гемоглобин, миоглобин, цитохром, пероксидазу, хромопротеид) или комплексы с натрием, калием, медью, ванадием, никелем, оловом, цинком, марганцем, кобальтом. При этом, механизм синтеза порфиринов одинаков в клетках всех тканей, однако скорость их образования/длительность существования существенно варьирует. Основной функцией порфиритовых комплексов является их участие в сложных метаболических процессах (транспортировка кислорода, биологическое окисление, фотосинтез и др.). Конечным продуктом пуринового метаболизма является мочевая кислота. Порфирины выделяются из организма с мочой, калом и желчью.
В целом порфириновая болезнь относятся к достаточно редким заболеваниям: обобщенный показатель заболеваемости различными формами болезни составляет 1:20 000 (Википедия). При этом, показатели заболеваемости на 100 тыс. населения широко варьирует — наследственная копропорфирия: 3-5 случаев; перемежающаяся острая перемежающаяся порфирия: в пределах 5-10 случаев; вариантная порфирия - 2-3 случая; поздняя кожная порфирия - 15-20 случаев на 100 тыс. населения. Порфирии не эндемичные заболевания (то есть характерные для определенной местности) и встречаются среди населения различных стран с одинаковой частотой.
Заболевание порфирия протекает как хронически, так и в виде острых атак. Существенно варьирует и возраст дебюта заболевания: эритропоэтические порфирии манифестируют преимущественно в 3-5 лет (дошкольном детстве), острые порфирии развиваются в 14-16 лет (период/после полового созревания), приобретенная спорадическая форма печеночной кожной порфирии — у лиц после 40 лет. Ввиду обширности темы рассмотрим лишь некоторые формы порфирий, в частности болезнь Гюнтера.
Патогенез всех форм порфирий имеет общие звенья, независимо от особенностей клинического течения/тканевой принадлежности. В их основе — отсутствие/снижение активности определённого фермента в общей цепи биосинтеза гема, что приводит к избыточному накоплению продукции в токсических концентрациях порфиринового обмена перед звеном, где локализуется дефектный энзим. Локализуются гены ферментов на разных хромосомах и групповой сцепленности не имеют.
Процесс синтезирование гема включает восемь последовательных этапов и каждый фермент отвечает свой этап и кодируется определенным геном. Соответственно каждая форма порфирии имеет свой специфичный ферментативный дефект. При хронических формах порфирий отмечается накопление копропорфирина, протопорфирина и уропорфирина; при острых формах — порфобилиногена/ДАЛК (дельта-аминолевулиновой кислоты).
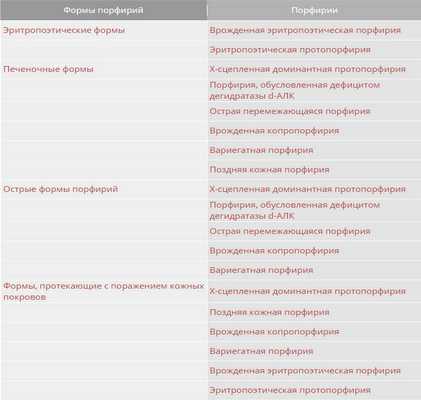
В основу квалификации порфирий положены такие признаки как место нарушения метаболизма порфиринов/накопления порфиринов и клинические проявления, согласно которым выделяют:
Соответственно, каждая из форм включает несколько видов порфирий. Обобщенная классификация форм и видов порфирий приведена в таблице ниже.
Причины
Причиной порфирий в подавляющем большинстве случаев являются мутации в генах, способствующие снижению активности фермента, принимающего участие в процессе биосинтеза гема. Наследуется заболевание преимущественно по аутосомно-доминантному, реже по аутосомно-рецессивному типу (болезнь Гюнтера). И только поздняя кожная порфирия (урокопропорфирия) может развиваться как вследствие длительной интоксикации тяжелыми металлами/заболеваний печени (опухоли печени, алкогольный/вирусный гепатит С), так и развивается при наличии наследственной предрасположенности.
Однако, для развития некоторых форм заболевания, в частности острая порфирия, кроме присутствия патологической мутации в гене фермента для манифестации симптоматики требуется воздействие провоцирующих факторов, которые стимулируют процесс выработки порфиринов. К такими общепризнанным факторам относятся сильные/постоянные стрессы, длительная инсоляция, злоупотребление алкоголем, голодание, инфекции бактериального/вирусного генеза, интоксикация тяжелыми металлами (ртуть, свинец), лекарственные средства (седативные средства, НПВС, сульфаниламиды, оральные контрацептивы, цефалоспорины, антималярийные препараты, барбитураты и др.), у женщин — менструальный цикл/беременность.
Наличие этих факторов способствует или повышенному потреблению гема, как конечного продукта или стимуляции активности начального фермента цикла биосинтеза. Как следствие — ускоряется синтез/накопление промежуточных продуктов обмена порфиринов в токсических концентрациях.
Симптомы
Клинические проявления порфирий варьируют в широких пределах и определяются конкретной формой заболевания. Рассмотрим лишь несколько форм заболевания.
Болезнь Гюнтера (врожденная эритропоэтическая порфирия)
Как и все другие эритропоэтические порфирии болезнь Гюнтера достаточно редкое явление. Манифестирует заболевание уже в раннем детстве (в возрасте 1-5 лет) и обусловлено врожденными ферментопатиями. Местом нарушения порфиринового обмена являются эритробласты костного мозга. Регистрируется в семьях исключительно среди сестер/братьев одного поколения и наследуется от любого из родителей как аутосомно-рецессивное. При этом, у родителей больных детей какие-либо клинические/биохимические признаки болезни отсутствуют. Для больных эритропоэтической порфирией характерен дефицит косинтетазы уропорфириногена, который концентрируется в патологической популяции эритробластов и в результате такой ферментативной блокады нарушается биосинтез гема, что приводит к накоплению в организме ребёнка критической концентрации уропорфирина I. Встречаются одинаково часто у лиц мужского/женского пола.
Для болезни Гюнтера характерны шесть основных признаков:
Манифестировать заболевание может как всеми признаками, так отдельными из них. Изменения со стороны кожи возникают преимущественно ранней весной на фоне сниженного диуреза и общей слабости. Проявляются изменения зудом/покраснением кожи на тыльной стороне кистей/стоп, ушных раковинах, лице, на голени и предплечьях после инсоляции, на месте которых позже появляются буллезные элементы (пузырьки) с серозно-геморрагическим содержимым. При этом кожа чрезвычайно чувствительна к механическим воздействиям. В случаях присоединении вторичной инфекции происходит изъязвление пузырьков и заживление с образованием рубцов.
Кожа неровная, кисти имеют когтеобразный вид, ногтевые пластинки утолщены/деформированы/помутневшие; в ряде случаев при хроническом течении разрушаются ушные раковины, отмечается мутиляция фаланг кисти с нарушением ее функции. На рентгенограмме остеопороз эпифизов фаланг, контрактура суставов (частичная/полная) в дистальных отделах конечностей. Для пациентов характерны длинные ресницы/густые брови, слабое физическое развитие, общее истощение, гиперпигментация, бледность кожи, окрашенные зубы, гипертрихоз на лице, увеличенная селезенка. Реже — помутнение хрусталика/роговицы. Лабораторно в кале/моче, плазме и в эритроцитах отмечается высокое содержание уро/копро/протопорфирина.
Поздняя кожная порфирия
Наиболее часто встречаемая порфирия печени, обусловленная нарушением процесса синтеза гемов печени, при которой отмечается повышенное образование/выделение копропорфирина/уропорфирина с мочой и их задержкой в кожных покровах. Урокопропорфирия манифестирует тремя основными дерматологическими симптомами: пигментация; пузыри; гипертрихоз.
Гиперпигментация кожи носит диффузный характер и возникает преимущественно на участках тела, подвергающихся солнечной инсоляции (кисти рук, шея, лицо, ушные раковины, верхняя часть груди). Цвет варьирует от красновато-синюшного (бронзового) до землисто-серого и определяется индивидуальными особенностями, наличием сопутствующих заболеваний и непосредственным влиянием внешних климатических/профессиональных факторов. Такая характерная реакция возникает лишь на местах облучения, и она расценивают как фотосенсибилизация (фотодерматоз), который при отсутствии облучения устраняется. Реакция усиливается летом, а зимой практически исчезает. По прошествии времени пигментация приобретает стойкий характер и становится более интенсивной, слабо изменяясь на протяжении года.
Следующим классическим признаком является пузырная реакция, которой предшествует повышенная ранимость кожи тыльной стороны кистей/лица. Проявляется образованием на видимо неизмененной коже пузырей размерами от просяного зерна до горошины округлой/овальной формы с жидкостным содержимым (вначале серозным, а затем мутнеет и переходит в гнойное). В дальнейшем пузыри самопроизвольно вскрываются, образуя эрозии с неправильными очертаниями с формированием впоследствии атрофических рубцов. Пузырная реакция увеличивается в весенне-летний период, а зимой пузыри, как правило, отсутствуют. Преимущественной локализацией пузырей являются кожа лица, шеи, ушных раковин, тыла кистей; реже — на волосистой части головы, предплечьях, губах. У женщин может иметь место атипичное расположение пузырей — на закрытых участках кожи: бедрах, спине, голенях. Длительность пузырной реакции варьирует от 1-2 недель до нескольких месяцев. В случаях присоединения вторичной инфекции отмечается лимфаденит/лимфангоит, глубокие язвы. Гипертрихоз - менее специфический синдром, встречается в среднем у 70% пациентов на лице в лобно-височной области.
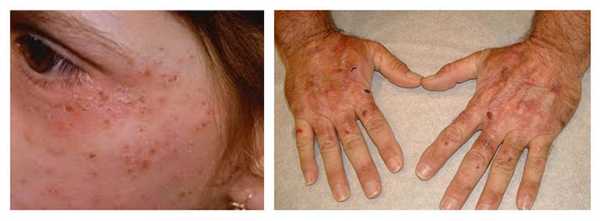
Для этой формы свойственны ожирение, нарушение функций ЖКТ, поражения печени, органа зрения: нарушение цветовосприятия, помутнение роговицы, конъюнктивит, расширение сосудов глазного дна. Кроме этих основных симптомов характерно преждевременное старение кожи и появление глубоких морщины. Ногти также подвергаются изменению: деформируются, теряют блеск, становятся матовыми; часто развивается подногтевой гиперкератоз. Пациенты выглядят старше своего возраста. Ниже приведены фото больных порфирией (поздняя кожная форма).
Острые порфирии: острая перемежающаяся порфирия
Встречается значительно чаще у лиц женского пола. В развитии заболевания существенную роль играют провоцирующие факторы. Для этой формы заболевания характерны несколько групп симптомов:
Клиническая симптоматика и ее выраженность определяется формой заболевания. Так, при скрытом/здоровом носительстве мутантного аллеля клинические признаки отсутствуют на фоне присутствия минимальных биохимических отклонений от нормы.
Анализы и диагностика
В целях диагностики используются специальные методы лабораторного определения порфиринов/их предшественников в кале и моче, и в крови — для оценки активности ферментов.
Так, для болезни Гюнтера в клинико-биохимических анализах крови характерны признаки гемолиза — повышение сывороточного железа/непрямого билирубина, пойкилоцитоз, ретикулоцитоз, сфероцитоз, анизоцитоз, увеличение печеночных трансаминаз. Для пациентов с острыми порфириями характерно снижение уровня натрия/глюкозы, при поздней кожной порфирии— увеличение ферритина/сывороточного железа, положительные маркеры на вирус гепатита С.
При постановке диагноза важен семейный анамнез: наличие у близких родственников заболевания, а также обстоятельства начала заболевания (беременность, менструации, инсоляция, прием алкоголя/лекарств, голодание, инфекции и др.). Для подтверждения диагноза определяются уровни различных ферментов цикла биосинтеза гема в плазме, эритроцитах, лимфоцитах.
Из инструментальных методов могут назначаться КТ/УЗИ органов брюшной полости, КТ/рентгенография грудной клетки, ЭКГ, ЭЭГ головного мозга и др.
Эффективные методы лечения, стойко корригирующие наследственную неполноценность ферментов и нарушения метаболизма порфиринов отсутствуют. Наиболее часто практикуют патогенетические методы лечения, облегчающие течение заболевания и в определенной степени предотвращающие развитие необратимых тяжелых изменений внутренних органов. В основе такой терапии выведение из организма накопившегося избыточного содержания порфиринов/метаболитов и устранение провоцирующих обострение неблагоприятных факторов. Способы лечения, медикаментозные препараты определяются конкретной формой порфирии.
Лечение эритропоэтических порфирий
Порфирии этой формы терапии поддаются очень плохо и сводится оно преимущественно к защите глаз/кожных покровов от солнечной инсоляции света, что достигается ношением закрытой одежды, нахождением в помещениях с остеклением не пропускающим УФ лучи, постоянным использованием фото защитных средств (кремов) с высокой степенью защиты (SPF 50+). Из препаратов: β-каротин, пероральный прием ежедневно в дозе 180-300 мг для взрослых /90-120 мг для детей; Уголь активированный, Никотиновая кислота, Цитохром. Показаны переливания свежей эритроцитарной массы, трансплантация костного мозга, спленэктомия. При эритропоэтической порфирии назначаются дополнительно гепатопротекторы (Аудеметионин, Урсодезоксихолевая кислота), Инозин, Аденозинмонофосфат, витамины В1, В2, В12, препараты железа; седативные средства, антигистаминные препараты, синтетические противомалярийные средства (Гидроксихлорохин, Хлорохин). Проводятся хронически перемежающиеся кровопускания.
Лечение острых порфирий
С целью купирования образования порфобилиногена назначают гема аргина (Нормосанг), гематин, производные АТФ (Фосфаден, Рибоксин, Аденил) и глюкозу в больших дозах. С целью купирования вегетативной симптоматики назначается Октреотид. Для восстановления в нервных волокнах миелина показаны витамины группы В. В случаях развития менструалозависимых атак назначают Золадекс, Синарел и другие агонисты гонадотропин-рилизинг гормона. Для купирования болевого синдрома при острой перемежающей порфирии назначают Нимесулид, Индометацин, Кетопрофен, салицилаты (Аспирин).
Лечение ПКП (поздней кожной порфирии)
Для удаления избытка железа/порфиринов проводится кровопускания (флеботомия), плазмаферез или назначаются комплексообразующие (Дефероксамин, Пентацин, Унитол, Тетацин-кальция, Дизфераль) и аминохинолиновые (Хлорохин) препараты. С целью снижения процесса всасывания в ЖКТ порфиринов назначаются энтеросорбенты (Активированный уголь); используются солнцезащитные кремы. При развитии гепатита С проводится противовирусная терапия (Рибавирин, Альфа-интерферон). Для нормализации функции печени и ЖКТ назначаются гепатопротекторы (Эссенциале форте), ферменты (Фестал, Панзинорм, Трифермент). Показаны витамины группы В, фолиевая кислота.
Читайте также: