Гемифациальная микросомия - лучевая диагностика
Добавил пользователь Валентин П. Обновлено: 29.01.2026
Синдром Тричер – Коллинза - Франческетти (мандибулофациальный дизостоз, TCOF, OMIM154500) - аутосомно-доминантное заболевание, характеризующееся нарушением черепно-лицевого развития. Клинические признаки: антимонголоидный разрез глаз, колобома (дефект) нижних век, микрогнатия (гипоплазия нижней челюсти), двусторонняя гипоплазия скуловых костей и орбит, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости. Часто встречается высокое арковидное нёбо или расщелина нёба, макростомия (большая ротовая щель), отсутствие ресниц на нижнем веке. В некоторых случаях отмечаются рост волос на щеках, колобомы верхнего века и радужки. У большинства пациентов слаборазвитые лицевые кости, что приводит к «затонувшему» лицу: крупный нос и очень маленькие челюсти и подбородок (микрогнатия). У некоторых больных присутствует волчья пасть. При тяжёлой патологии микрогнатия может вытеснять язык у новорождённых, создавая опасную преграду в ротоглотке для дыхания.
Для заболевания характерна высокая пенетрантность (т.е. высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность (т.е. различный характер и тяжесть проявления болезни). Молекулярно-генетической причиной заболевания являются, как правило, нонсенс мутации в генеTCOF1, приводящие к возникновению преждевременного стоп кодона и, как следствие, к гаплонедостаточности (состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма). Продукт гена - ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. Возможна прямая молекулярно-генетическая диагностика этого синдрома, которая заключается в выявлении изменений нуклеотидной последовательности в гене TCOF1 методом прямого автоматического секвенирования.
Аутосомно-доминантный. Для заболевания характерна высокая пенетрантность (т.е. высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность (т.е. различный характер и тяжесть проявления болезни).
Мутацию в гене TCOF1 можно выявить в 78%-93% случаев, в 8% выявляют мутации в генах POLR1C или POLR1D.
Продукт гена - ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. У больных весьма характерное лицо. Клинические признаки: антимонголоидный разрез глаз, колобома (дефект) нижних век, микрогнатия (гипоплазия нижней челюсти), двусторонняя гипоплазия скуловых костей и орбит, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости. Часто встречается высокое арковидное небо или расщелина неба, макростомия (большая ротовая щель), отсутствие ресниц на нижнем веке. В некоторых случаях отмечаются рост волос на щеках, колобомы верхнего века и радужки. Порок развития уха заключается в деформации ушной раковины, отсутствии костного отдела наружного слухового прохода, недоразвитии барабанной полости и слуховых косточек. Отмечаются также гипоплазия больших пальцев лучевой и локтевой костей, расщелины неба. Тугоухость смешанного характера с одновременным поражением звукопроведения и звуковосприятия.
Интеллект, как правило, не страдает. Трудности с дыханием и питанием могут возникнуть в первые годы из-за стеноза верхних дыхательных путей и ограниченного открытия рта.
Анализ симптомокомплексов у пациентов с синдромом краниофациальной микросомии и их лечение
Актуальность. Краниофациальная микросомия – собирательное определение, объединяющее врожденные патологии органов, развивающихся из I и II жаберных дуг. Однако принадлежность различных врожденных патологий к данному заболеванию остается спорной. По этой причине нет стандартизированных показаний к срокам и методам лечения.
Материалы и методы. В данной работе проанализированы результаты обследований 89 детей и подростков от 1 до 18 лет с синдромами краниофациальной микросомии, проведенных с 2011 по 2021 год. Результаты. Пациенты разделены на группы по степени тяжести их патологии и возрасту. В зависимости от варианта фенотипа, были предложены различные методы лечения.
Выводы. На основании нашего и мирового опыта, а также учитывая анатомические и функциональные изменения у детей и подростков с синдромами краниофациальной микросомии, возникает актуальность создания схемы для построения индивидуальных, мультидисфиплинарных алгоритмов лечения данной категории пациентов.
Ключевые слова
Об авторах
Московский государственный медико-стоматологический университет им. А.И. Евдокимова; Медицинский университет «Реавиз», филиал
Россия
Имшенецкая Наталья Ильинична, кандидат медицинских наук, ассистент кафедры детской челюстно-лицевой хирургии; доцент кафедры стоматологии
Топольницкий Орест Зиновьевич, доктор медицинских наук, профессор, заслуженный врач России, заведующий кафедрой детской челюстно-лицевой хирургии
Смыслёнова Маргарита Витальевна, доктор медицинских наук, профессор кафедры лучевой диагностики
Московский государственный медико-стоматологический университет им. А.И. Евдокимова; Российская медицинская академия непрерывного профессионального образования
Россия
Лежнев Дмитрий Анатольевич, доктор медицинских наук, профессор, заведующий кафедрой лучевой диагностики; профессор кафедры терапевтической стоматологии
Медицинский университет «Реавиз», филиал; Московский государственный медико-стоматологический университет им. А.И. Евдокимова
Россия
Слюсар Ольга Ивановна, кандидат фармакологических наук, декан факультета непрерывного медицинского образования, доцент кафедры фармакологии
Список литературы
2. Grabb WC. The first and second branchial arch syndrome. Plast Reconstr Surg November. 1965;36:485–508. doi: 10.1097/00006534-196511000-00001
4. Bennun RD, Mulliken JB, Kaban LB, Leonard BD, Murray JE. Microtia: a microform of hemifacial microsomia. Plast Reconstr Surg. 1985;76(6):859-863. doi: 10.1097/00006534-198512000-00010.
5. Rollnick BR, Kaye CI, Opitz JM. Hemifacial microsomia andvariants: pedigree data. Am J Med Genet. 1983;15(2):233-253. doi: 10.1002/ajmg.1320150207.5
7. Stark RB, Saunders DE. The first branchial syndrome. The oral-mandibular-auricular syndrome. Plast Reconstr Surg Transplant Bull. 1962;29:229–239. doi: 10.1097/00006534-196203000-00001
11. Birgfelt C, Heike C. Craniofacial Microsomia. Clin Plastic Surg 2019;46(2);207–221. doi: 10.1016/j.cps.2018.12.001
13. Caron CJJM, Pluijmers BI, Wolvius EB, Looman CWN, Bulstrode N, Evans RD, et al. Craniofacial and extracraniofacial anomalies in craniofacial microsomia: a multicenter study of 755 patients. J Craniomaxillofac Surg. 2017;45(8):1302–1310. doi: 10.1016/j.jcms.2017.06.001/
14. Konas E, Canter HI, Mavili ME. Goldenhar complex with atypical associated anomalies: is the spectrum still widening? J Craniofac Surg. 2006;17(4):669–672. doi: 10.1097/00001665-200607000-00011
15. Tuin J, Tahiri Y, Paliga JT, Taylor JA, Scott P Bartlett SP. Distinguishing Goldenhar syndrome from craniofacial microsomia. J Craniofac Surg. 2015;26(6):1887-1892. doi: 10.1097/SCS.0000000000002017
16. Caron CJJM, Pluijmers BI, Maas BDPJ, Klazen YP, Katz ES, Abel F, et al. Obstructive sleep apnoea in craniofacial microsomia: analysis of 755 patients. Int J Oral Maxillofac Surg. 2017;46(10):1330–1337. doi: 10.1016/j.ijom.2017.05.020
17. van de Lande LS, Caron CJJM, Pluijmers BI, Joosten KFM, Streppel M, Dunaway DJ, et al. Evaluation of swallow function in patients with craniofacial microsomia: a retrospective study. Dysphagia. 2018;33:234–242. doi: 10.1007/s00455-017-9851-x
18. Rajendran T, Ramalinggam G, Kamaru Ambu V. Rare presentation of bilobed posterior tongue in Goldenhar syndrome. BMJ Case Rep. bcr-2017-219726. doi: 10.1136/bcr-2017-219726
19. Chen EH, Reid RR, Chike-Obi C, et al. Tongue dysmorphology in craniofacial microsomia. Plast Reconstr Surg 2009;124:583–589. doi: 10.1097/PRS.0b013e3181addba9
20. Birgfeld CB, Heike CL, Saltzman BS, Leroux BG,Evans KN, Luquetti DV. Reliable classification of facial phenotypic variation in craniofacial microsomia: a comparison of physical exam and photographs. Head Face Med. 2016; 12:14. doi: 10.1186/s13005-016-0109-x.
21. Allam AK. Hemifacial Microsomia: Clinical Features and Associated Anomalies. J Craniofac Surg. 2021;32(4):1483-1486. doi: 10.1097/SCS.0000000000007408
Врожденные деформации
Cинонимы: синдром первой и второй жаберных дуг, синдром Гольденхара – окуло-ауриколо-вертебральная дисплазия, краниофациальная микросомия, отомандибулярный дизостоз и латеральная фациальная дисплазия - является редким наследственным заболеванием, характеризующимся значительным числом аномалий, которые возникают вследствие нарушения развития первой и второй жаберных дуг первого глоточного кармана, первой жаберной щели и зачатков височной кости. Среди врожденных пороков развития черепно-челюстно-лицевой области гемифациальная микросомия занимает второе место по частоте встречаемости после врожденных расщелин верхней губы и нёба. Частота этого синдрома колеблется в пределах 1:3500-5600 новорожденных, он присутствует у 1 из 1000 у детей с врожденной глухотой. Распределение по половому признаку (мужчины и женщины) составляет примерно 3:2.
Этиология и тип наследования изучены недостаточно. Неблагоприятный акушерско-гинекологический анамнез матери (предшествующие аборты, сахарный диабет, избыточный вес) и тератогенные факторы на ранних сроках беременности являются отягощающими факторами риска рождения больного ребенка. Вероятность повторного рождения больного ребенка, как и вероятность рождения больного ребенка у носителя патологии, ориентировочно равна 2%.
Клинически для гемифациальной микросомии характерны: недоразвитие тела и ветви нижней челюсти, гипоплазия скуловой кости и дуги, недоразвитие структур ВНЧС; аплазия ветви нижней челюсти и ВНЧС; нарушение размеров и положения глазницы; гипоплазия, аплазия ушной раковины, атрезия слухового прохода, поражение лицевого нерва, гипоплазия мимических мышц; дефицит мягких тканей; макростомия; предушные придатки и свищи; иногда сочетание с врожденной расщелиной губы и неба, эпибульбарным дермоидом, аномалией прикуса, адентией, нарушением структуры эмали и формы зубов, пороками развития опорно-двигательного аппарата, органов зрения и нервной системы, а также аномалиями мочевыделительной системы и желудочно-кишечного тракта.
Cклеродермия
Это прогрессирующее системное заболевание, в основе которого лежит воспалительное поражение мелких сосудов всего организма, с последующими фиброзно-склеротическими изменениями кожи, опорно-двигательного аппарата и внутренних органов.
При очаговой склеродермии наблюдается ограниченное уплотнение кожи, но могут вовлекаться подкожные ткани и кости. Выделяют две основные формы очаговой склеродермии – бляшечную (морфея) и линейную. В первом случае поражение кожи имеет вид уплотнений округлой формы («Бляшки»), с лиловым ободком по периферии в дебюте болезни. Эти очаги, единичные или множественные, могут появляться как на туловище, так и на лице и конечностях. При линейной форме очаговой склеродермии участки поражения имеют вид полос уплотнения кожи, часто с вовлечением подлежащих мышц и костей, и локализуются, главным образом, на конечностях и лице. Эта форма очаговой склеродермии в случае развития в детском и подростковом возрасте, может приводить к ограничению движений (мышечные и суставные контрактуры) и нарушениям развития пораженных участков. Внутренние органы при очаговой склеродермии не страдают.
Системная склеродермия (ССД) – форма склеродермии, при которой помимо уплотнений кожи развиваются разнообразные поражения суставов, внутренних органов (сердца, легких, желудочно-кишечного тракта, почек). В редких случаях наблюдается поражение только внутренних органов, без изменений кожи. Женщины заболевают в 3-5 раза чаще, чем мужчины. ССД подразделяется на лимитированную и диффузную форму, которые различаются по распространенности и выраженности поражения кожи и внутренних органов.
Липодистрофия
Общее или локальное поражение подкожной клетчатки с уменьшением (атрофическая форма) или увеличением (гипертрофическая форма) объема жировой ткани. Л. могут быть генерализованными или сегментарными. К липодистрофиям относят следующие патологические состояния: врожденную генерализованную Л., гипермускулярную Л., прогрессирующую сегментарную Л., или болезнь Барракера — Симонса, постинъекционную Л., Липоматоз болезненный (болезнь Деркума).
Прогрессирующая сегментарная липодистрофия (синонимы липоатрофия, болезнь Барракера-Симона) — это Относительно редкое заболевание, проявляющееся атрофией подкожной жировой клетчатки лица, шеи, плечевого пояса, грудной клетки при нормальном или избыточном отложении жира в нижней половине тела. Встречается редко, поражает преимущественно женщин. Этиология неясна. Внутренние органы не поражаются, функция их не нарушена. Больные обращаются к врачу из-за своей внешности, иногда жалуются на слабость, раздражительность. Жизненный прогноз благоприятен. Специфичного лечения нет, применяют средства, укрепляющие нервную систему, и витамины.
Микрогения
Это аномалия развития лица, характеризующееся гипоплазией (недоразвитием) нижней челюсти.
В основе деформации лежит нарушение роста и развития нижней челюсти в результате травмы или патологического процесса в области сустава и ветви челюсти, перенесенных во время родов, в детском возрасте при незаконченном росте лицевого скелета, а также вследствие врожденных нарушений во время развития нижне- и верхнечелюстного отростков головной части зародыша. При односторонней микрогении лицо асимметрично за счет укорочения ветви и горизонтальной части тела челюсти, что приводит к смещению подбородка в сторону укорочения. Пораженная сторона выглядит более выпуклой, здоровая сторона уплощена, тело челюсти удлинено и деформировано. Двусторонняя (симметричная) микрогения характеризуется укорочением тела и ветвей челюсти, что сопровождается смещением подбородочного отдела кзади и его «скошенностью». При этом верхняя челюсть выступает вперед, придавая лицу так называемое «птичье выражение». Деформация нижней челюсти может быть выражена в различной степени: от незначительной асимметрии или уплощения подбородочного отдела с умеренным нарушением прикуса до тяжелых, обезображивающих деформаций. При выраженном нарушении прикуса развиваются функциональные расстройства из-за затруднения акта откусывания и пережевывания пищи.
Микрогения довольно часто сочетается с анкилозом височно-челюстного сустава.
Гемифациальная микросомия - лучевая диагностика
Высокие технологии в помощь детям
Детская городская клиническая больница св. Владимира – одна из крупнейших в Москве. Это многопрофильное учреждение в 2016 году отметит свое 140-летие. Ежегодно за госпитализацией сюда обращается около 20 тысяч человек, выполняется около 8 тысяч операций различной сложности. Кроме того, консультацию специалистов и амбулаторное лечение получают около 19 тысяч человек. В больнице установлено самое современное оборудование, позволяющее проводить томографию, звуковые и эндоскопические исследования. Лечение детей в ДГКБ св. Владимира предполагает тесное взаимодействие лечащих врачей между собой, гарантируя 100%-ую точность поставленного диагноза и использование правильной программы лечения
Главный врач ГБУЗ «Детская городская клиническая больница святого Владимира Департамента здравоохранения города Москвы», доктор медицинских наук Владимир Попов
Отделение челюстно-лицевой хирургии
В течение вот уже 25 лет здесь оказывается высококвалифицированная специализированная медицинская помощь детям с экстренной и плановой хирургической патологией в челюстно-лицевой области.
Отделение является ответственным учреждением города Москвы по оказанию специализированной хирургической помощи детям с травмами и воспалительными заболеваниями челюстно-лицевой области. С каждым годом количество обращений пациентов с патологией челюстно-лицевой области неуклонно увеличивается. Ежегодно отмечается рост числа детей с воспалительными процессами на 15–16%. Из года в год растет детский травматизм. Детей с травмой мягких тканей за последние 5 лет увеличилось на 75%, с переломами костей лицевого скелета на 58%. Общее количество больных детей за 2014 год, которым была оказана специализированная медицинская помощь, составило 7359 человек: 6146 в стационаре и 1213 в консультативно-диагностическом отделении. Оперативная активность возросла на 23%, а количество сложных плановых операций увеличилось на 15%. Также врачи отделения ведут интенсивный консультативный прием.
Заведующий отделением челюстно-лицевой хирургии, доктор медицинских наук, профессор кафедры стоматологии детского возраста и ортодонтии Первого Московского государственного медицинского университета им. И.М. Сеченова Дмитрий Юрьевич Комелягин. В отделении работают 12 врачей: 7 челюстно-лицевых хирургов, нейрохирург, микрососудистый хирург, специалист по лучевой диагностике, ортодонт, педиатр. Все врачи имеют высшую квалификационную категорию. Из них 7 являются кандидатами медицинских наук.
Активно ведется научно-клиническая работа с внедрением ее результатов в практику. Коллектив обладает 8 патентами на изобретения и методы. Сотрудники регулярно участвуют в научно-практических конференциях национального и международного уровней. Специалисты отделения часто выезжают для оказания специализированной медицинской помощи детям со сложными переломами челюстно-лицевой области в другие ЛПУ Москвы: ДГКБ № 1, 7, 9.
Также здесь проводится лечение детей с врожденной патологией в челюстно-лицевой области:
– расщелины губы и неба, альвеолярного отростка верхней челюсти;
– пороки развития черепа и лицевого скелета (синдромы Крузона, Апера, Пфейффера, Пьера Робена, Тричера-Коллинза, Нагера, Ханхарта, Халлермана-Штрайффа-Франсуа, гемифациальная микросомия и другие челюстно-лицевые дизостозы);
– краниосиностозы (плагиоцефалия, тригоноцефалия, брахицефалия);
– артрозы и анкилозы височно-нижнечелюстного сустава;
– опухолеподобные образования и кисты челюстей;
– дефекты и деформации ушных раковин;
– посттравматические деформации костей и мягких тканей;
– срединные и боковые кисты и свищи шеи;
– сосудистые образования головы и шеи (младенческие гемангиомы, гемангиоперицитомы, гемангиоэндотелиомы; лимфатические, венозные, артериальные, артериовенозные, капиллярные мальформации).
Отделение является единственным в России, в котором проводится хирургическое лечение новорожденных с синдромом Пьера Робена с применением компрессионно-дистракционного остеосинтеза. Опыт лечения такой патологии 14 лет, имеется 1 патент на изобретение, аппараты и методы собственной разработки.
В практику отделения челюстно-лицевой хирургии активно внедряются методы эндоскопической хирургии. В 2012 году получен патент на изобретение № 2455952 «Способ лечения переломов мыщелкового отростка нижней челюсти со смещением под основание черепа с использованием эндоскопической техники».
Высокотехнологичные методы обследования и лечения
Мы оказываем высокотехнологичную помощь с использованием методов биомоделирования. При использовании биомоделирования врач получает возможность иметь точную твердотельную копию структур, которые в обычных условиях были бы недоступны для осмотра, визуального и тактильного восприятия.
Пластиковые модели предоставляют хирургу уникальную возможность спланировать и отработать технику будущей операции; более точной диагностики и изготовления эндопротезов для устранения послеоперационных дефектов и деформаций. При устранении дефекта одной из сторон симметричного объекта биомоделирование дает дополнительную возможность построить зеркальную модель на основе здоровой половины и по ней изготовить эндопротез, который получается полностью симметричным противоположной здоровой стороне. Использование биомоделирования позволяет сократить время операций на 20–50%. Таким образом, выявление и учет индивидуальных особенностей пациента при планировании и проведении операций ведет к повышению качества лечения и сокращению сроков реабилитации больного, что в целом положительно сказывается на экономическом состоянии государства.
Работа отделения была высоко оценена Швейцарско-российским форумом в 2014 году, 8 декабря коллективу вручена премия им. А.В. Суворова за самый выдающийся инновационный проект года – 3DMedBioprinting.
В настоящее время отмечается увеличение обращений пациентов с сосудистыми образованиями головы и шеи. С начала 2011 года в отделении начато лечение детей с гемангиомами, лимфангиомами и сосудистыми мальформациями головы и шеи согласно международным стандартам InternationalSocietyfortheStudyofVascularAnomalies(ISSVA). На сегодняшний момент обследовано и находится на лечении более 450 пациентов с обширными комбинированными гемангиомами головы и шеи. Пролечено 350 пациентов, получены хорошие косметические и функциональные результаты. Кроме того, активно внедряются в практику современные высокотехнологичные малоинвазивные хирургические методы лечения обширных лимфангиом головы и шеи с применением ультразвуковой навигации.
Ежегодно в отделении проходят обследование и лечение более 7000 детей не только из Москвы и Московской области, но и из всех регионов России, ближнего и дальнего зарубежья. Доля пациентов с врожденной и приобретенной патологией составляет 40%. В настоящее время выполняются все основные операции, известные в мировой практике, без ограничения по возрасту детей. Современный технический прогресс позволяет внедрять в медицинскую практику все новые и новые высокотехнологичные методы обследования и лечения, благодаря которым значительно повышается эффективность лечения детей со сложной врожденной и приобретенной патологией черепно-челюстно-лицевой области.
Все это возможно благодаря тому, что отделение работает в условиях многопрофильного стационара с наличием всех необходимых смежных специалистов, что обеспечивает комплексный мультидисциплинарный подход к лечению детей со сложной черепно-челюстно-лицевой патологией, а также в тесном сотрудничестве с научными кафедрами: детской хирургии РМАПО, педиатрии МГМСУ им. А.И. Евдокимова, ЛОР-болезней РМАПО, стоматологии детского возраста и ортодонтии ПМГМУ им. И.М. Сеченова.
Первый опыт лапароскопических реконструктивных операций на поджелудочной железе
Хирургия поджелудочной железы (ПЖ) является одним из самых сложных разделов абдоминальной хирургии у детей. Ретроперитонеальное расположение органа, близость к 12-перстной кишке и крупным висцеральным сосудам, необходимость в наложении панкреатодигестивных анастомозов делают хирургические вмешательства на ПЖ технически сложными и трудно воспроизводимыми.
В последние годы в детской хирургии широкое распространение получили мини-инвазивные вмешательства. В ДГКБ святого Владимира лапароскопические операции по поводу аномалий и заболеваний ПЖ были выполнены 33 больным в возрасте от 1 месяца до 16 лет. Эктопированная в желудок ПЖ (хористома) имелась у 16 детей, кольцевидная ПЖ – у 2, посттравматическая или постнекротическая киста ПЖ – у 6, ретенционная киста ПЖ – у 2, эхинококковая киста головки ПЖ – у 1, киста вирсунгова протока – у 1, врожденное расширение вирсунгова протока и вирсунголитиаз – у 1, удвоение желудка с локализацией в ПЖ – у 2, солидно-псевдопапиллярная опухоль ПЖ – у 2 детей.
Были выполнены следующие лапароскопические вмешательства: наложение дуодено-дуоденоанастомоза по Кимура, гастротомия и иссечение хористомы желудка, экстирпация из ПЖ кистозных удвоений желудка, наружное дренирование посттравматических кист ПЖ, иссечение врожденных кист ПЖ, эхинококкэктомия из головки ПЖ, панкреатоцистоеюноанастомоз с выключенной по Ру петлей тонкой кишки, дистальный панкреатоеюноанастомоз по Ру, продольный панкреатикоеюноанастомоз по Фрею, дистальная резекция ПЖ, секторальная резекция ПЖ с наложением панкреатогастроанастомоза.
Приводим два клинических наблюдения успешных лапароскопических реконструктивных операций на ПЖ у детей
Клиническое наблюдение 1. Больной К. 7 лет, поступил в отделение абдоминальной эндоскопической хирургии ГДКБ святого Владимира для обследования и определения дальнейшей тактики лечения. Из анамнеза: 4 месяца назад по месту жительства перенес операцию: лапаротомию, дренирование сальниковой сумки по поводу разрыва тела ПЖ. После удаления дренажа сформировалась киста ПЖ, которая имела тенденцию к увеличению. При поступлении проведено УЗИ органов брюшной полости: в проекции тела ПЖ определяется кистозное образование диаметром 48 мм, расцененное как ложная киста поджелудочной железы. С целью дифференциальной диагностики выполнена спиральная компьютерная томография брюшной полости, результаты которой позволили подтвердить диагноз и определить показания к операции.
31 марта 2015 годы был оперирован, выполнены лапароскопическое иссечение кисты поджелудочной железы, наложение дистального панкреатоеюноанастомоза по Ру, дренирование сальниковой сумки. При лапароскопии установлено, что в брюшной полости умеренно выраженный спаечный процесс. Острым и тупым путем от передней брюшной стенки отделен сальник и поперечная ободочная кишка. Область сальниковой сумки занята объемным кистозным образованием до 6 см в диаметре. Через корень брыжейки ободочной кишки киста продольно вскрыта с помощью электрокаутера – под давлением выделился светлый панкреатический секрет. С помощью электрокаутера Martinпроизведено иссечение стенок кисты с сохранением фиброзных тканей на культе головки ПЖ. Имеется полный поперечный разрыв ПЖ на уровне перешейка. Тело ПЖ частично мобилизовано влево с отведением верхней брыжеечной вены и селезеночных сосудов. В 20 см от связки Трейтца тонкая кишка поперечно пересечена сшивающим аппаратом Autosuter45. Дистальный конец проведен позадиободочно в сальниковую сумку. Наложен инвагинационный дистальный панкреатоеюноанастомоз «конец в конец» двухрядным швом нитями PDSII5-0. В 30 см дистальнее аборального конца с помощью аппарата Autosuter45 наложен межкишечный анастомоз «бок в бок». Места введения бранш сшивающего аппарата ушиты обвивным швом нитями викрил 4-0. Сальниковая сумка дренирована трубчатым дренажом слева через троакарный доступ.
Послеоперационный период протекал гладко, дренаж удален на 7 сутки. При контрольном УЗИ органов брюшной полости остаточных полостей не выявлено. Микроскопическое исследование препарата – посттравматическая киста ПЖ с воспалительными изменениями стенки. Выписан домой в удовлетворительном состоянии.
Клиническое наблюдение 2.Больной М. 5 лет, поступил в отделение абдоминальной эндоскопической хирургии ГДКБ святого Владимира. Из анамнеза известно, что в марте 2015 года появились резкие боли в животе, по поводу чего был обследован амбулаторно, заподозрена дилатация панкреатического протока. При УЗИ: ПЖ увеличена, размерами 15 × 11 × 12 мм, паренхима истончена, гетерогенная, без очаговых деструктивных проявлений. Вирсунгов проток расширен до 8 мм, в просвете фиксированные включения, эхографически расцененные как конкременты средней эхогенности, без акустической тени. Выполнена магнитно-резонансная холангиопакреатография, результаты которой подтвердили эхографические находки.
15 мая 2015 года больной был оперирован: выполнено лапароскопическое наложение продольного панкреатикоеюноанастомоза с выключенной петлей по Ру. В ходе лапароскопии с помощью коагулятора EnSealразделена желудочно-ободочная связка, широко вскрыта сальниковая сумка. Двумя транспариетальными швами желудок фиксирован к передней брюшной стенке. При ревизии поджелудочная железа увеличена, крупнобугристая на всем протяжении. С помощью монополярной коагуляции в области тела железы вскрыт пакреатический проток – выделился пакреатический сок со взвесью. Пакреатический проток продольно рассечен от хвоста до головки железы. Отмечается расширение протока до 1 см, в просвете имеется множество рыхлой консистенции панкреатических конкрементов до 0,5–1 см, которые удалены (рис. 5). Начальный участок тощей кишки экстраперитонизирован через расширенное до 2,5 см околопупочное троакарное отверстие. Кишка поперечно пересечена сшивающим аппаратом Autosuter45, наложен межкишечный анастомоз «бок в бок» однорядным обвивным швом нитью викрил 5-0. Кишка погружена в брюшную полость, вновь наложен карбоксиперитонеум. Ру-петля проведена позадиободочно и продольно рассечена по противобрыжеечному краю. Наложен продольный пакреатикоеюноанастомоз «бок в бок» с захватом головки железы обвивным швом нитью викрил 4-0. Сальниковая сумка дренирована трубчатым дренажом слева через контрапертуру в люмбо-дорзальной области.
Послеоперационный период протекал гладко. При контрольном УЗИ органов брюшной полости остаточных полостей не выявлено. Заживление ран первичное. Ребенок выписан домой в удовлетворительном состоянии.
Таким образом, лапароскопические вмешательства могут быть применены у детей с разнообразными аномалиями и заболеваниями ПЖ. Объем и особенности техники оперативных вмешательств определяются видом патологии ПЖ, возрастом больных и развившимися осложнениями.
Микрогнатия - симптомы и лечение
Что такое микрогнатия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Сенюка Андрея Николаевича, челюстно-лицевого хирурга со стажем в 17 лет.
Над статьей доктора Сенюка Андрея Николаевича работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Микрогнатия — это недоразвитие нижней или верхней челюстной кости.
Данный термин берёт своё начало от древнегреческих слов " μικρός" – "малый" и "γνάθος" — " челюсть" [1] . Он представляет собой лишь описательную характеристику и не является конкретным диагнозом. При его обсуждении правильнее говорить о зубочелюстных деформациях или различных видах дисгнатии, при которых отмечается недоразвитие (микрогнатия) какой-то части лицевого скелета или челюсти. Например, термин "дисгнатия" охватывает следующие нарушения прикуса:
- микрогнатию нижней челюсти с увеличенной, нормальной или уменьшенной верхней челюстью — дисгнатия II класса;
- микрогнатию верхней челюсти с нормальной или увеличенной нижней челюстью — дисгнатия III класса.
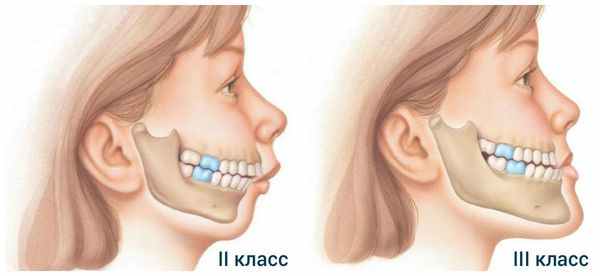
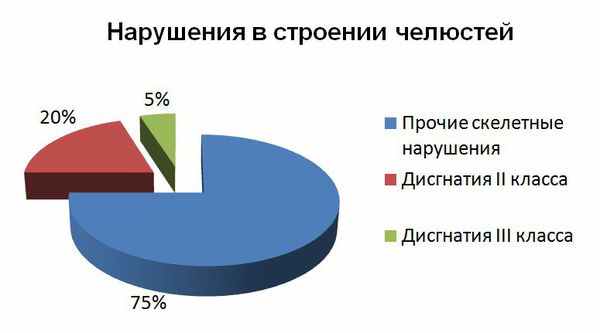
Казалось бы, данные нарушения в строении челюстей встречаются не так часто, однако практически каждый неправильный прикус, с которым родители маленьких пациентов обращаются к ортодонтам, связан именно со скелетными нарушениями роста челюстей. Современные данные указывают на распространённость микрогнатии: дисгнатия II класса встречается почти в 20 % случаев, дисгнатия III класса — в 5 % случаев [13] .
В зависимости от типа дисгнатии её причинами могут быть как генетические факторы (дисгнатия II и III классов, синдромальные микрогнатии), так и совокупность генетических и средовых факторов (дисгнатии II класса), влияющих на потенциал роста нижней челюсти [2] [3] [4] .
Многие факторы оказывают влияние на ребёнка ещё в утробе матери. Процесс формирования костей, в том числе и челюстных, у плода протекает с разной интенсивностью и в разное время: в один момент верхняя челюсть может оказаться меньше нижней, в другой момент — наоборот.
Спровоцировать недоразвитие челюсти у плода могут следующие причины:
- скудное неполноценное питание, например, лишённое кальция и других необходимых микроэлементов;
- вредные привычки матери — употребление алкоголя, наркотиков;
- инфекционные заболевания, перенесённые женщиной во время беременности.
В связи с этим микрогнатия часто проявляется у детей ещё во младенческом возрасте. Из-за недоразвития челюсти у младенцев могут наблюдаться сложности с приёмом пищи и затруднённое дыхание. Также микрогнатия возникает и после родов: ребёнок может приобрести или усугубить имеющийся дефект.
Среди основных факторов, влияющих на развитие микрогнатии, выделяют следующие:
- нарушенное сосание у младенца — неестественный захват груди, сóска неанатомической формы, длительное сосание пальцев приводят к наклону зубов вперёд и формированию открытого прикуса;
- поражения молочных моляров — ранняя потеря молочных жевательных зубов ведёт к недоразвитию челюстных костей из-за снижения активности зон роста;
- задержка прорезывания постоянных зубов;
- различные заболевания, вызывающие отклонения в процессе формирования костей, — рахит, тяжёлые инфекционные или эндокринные заболевания, остеомиелит;
- врождённые аномалии развития челюстныйх костей: синдром Марфана, гемифациальная микросомия и др.;
- патологии дыхательных органов — из-за неправильного строения носовой перегородки или хронических з аболеваний носоглотки человек начинает дышать через рот, что меняет тонус круговой мышцы рта и сужает верхнюю челюсть;
- травмы лицевой части головы и др.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы микрогнатии
Практически каждый тип дисгнатии и микрогнатии сопровождается вполне конкретными и заметными функционально-эстетическими нарушениями. К ним относятся все случаи неправильного прикуса: от мезиального, когда нижние зубы выступают вперёд относительно верхних, до любых вариантов дистального прикуса, при которых нижний зубной ряд находится намного позади верхнего ряда. Такие нарушения прикуса всегда сопровождаются определёнными негативными проявлениями:
- со стороны зубов: сти раемостью, кариозными поражениями , заболеваниями пародонта;
- со стороны зубных рядов: скученностью и компенсаторными нарушениями (сужением зубного ряда и его искривлением);
- со стороны височно-нижнечелюстных суставов: болью, щелчками, крепитацией (хрустящим звуком), ограничениями движений.
При скелетных нарушениях, связанных с уменьшением верхних дыхательных пространств из-за малых размеров опорных структур верхней и нижней челюстей, частым проявлением патологии являются синдромы обструкции дыхания во сне, например сонное апноэ.
Синдром обструктивного апноэ сна — это потенциально опасное для жизни состояние. Оно характеризуется храпом и периодически повторяющимися циклами прекращения дыхания во время сна. Чаще всего патология связана с сужением дыхательного пространства, вызванным неправильным прикусом и положением челюстных костей. Помимо храпа с остановками дыхания и ночных болей в грудной области сонное апноэ сопровождается снижением уровня кислорода в крови, грубо фрагментированным сном, избыточной дневной сонливостью, мигренями и раздражительностью.
Несмотря на перечисленные функциональные нарушения, самым значимым клиническим проявлением дисгнатий и микрогнатий является эстетический компонент. Именно неудовлетворённость улыбкой и лицом заставляют пациентов обращаться за медицинской помощью. Если для исправления улыбки часто бывает достаточно вмешательства ортодонта и/или стоматолога-ортопеда, то для восстановления эстетического баланса лица необходима помощь челюстно-лицевого хирурга [11] .
Основные признаки сильного недоразвития челюсти обычно можно увидеть невооружённым глазом. К ним относятся:
- непропорциональные черты, искажение контуров лица;
- неправильный прикус;
- западание верхней или нижней губы;
- скошенный ("птичий") или выступающий вперёд подбородок;
- сморщенная кожа подбородка и др.
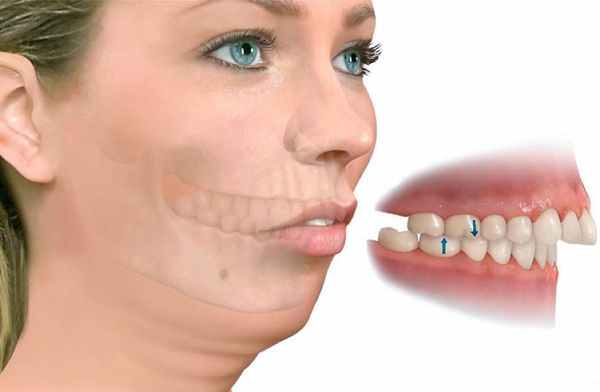
Выраженная микрогнатия хорошо заметна со стороны, но на ранних этапах диагностировать недоразвитие челюстных костей может только врач.
Патогенез микрогнатии
Ребёнок наследует от отца с матерью особенности анатомии всей зубочелюстной системы: строение и размер зубов, челюстей, мышц [12] . Иногда размер и форма зубов наследуется от одного родителя, а строение челюстей — от другого. Это может стать причиной нарушения гармоничного соотношения челюстей и зубных рядов.
Наследственные заболевания также служат причино й патологических изменений в строении лицевого скелета. Особую роль играют заболевания матери: они напрямую влияют на формирование жевательно-речевого аппарата, м огут нарушать его.
Однако генетика — не единственна я возможная причина недоразвития костей челюсти. Например, пребывание будущих родителей или новорождённого в неблагоприятной экологической обстановке (ультрафиолетовое облучение, повышенная радиация, содержание в воздухе и воде вредных элементов) или приём лекарств беременной женщиной могут стать главной или второстепенной причиной возникновения той или иной зубочелюстной аномалии.
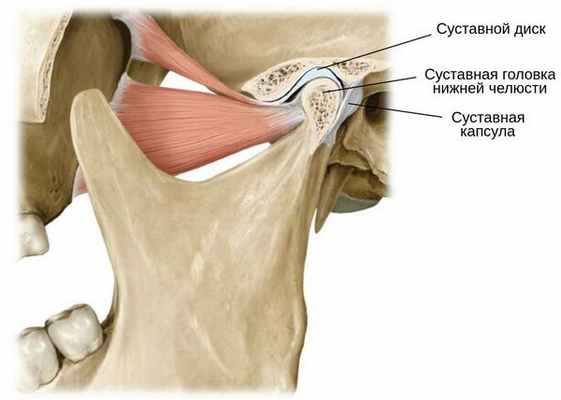
Основной потенциал роста нижней челюсти, который также стимулирует рост верхней челюсти, находится в ростковой зоне — хрящевом покрытии суставной головки нижней челюсти. Поэтому любые нарушения височно-нижнечелюстных суставов (смещение внутрисуставного диска), гормонозависимое и идиопатическое рассасывание суставной головки либо тормозят рост нижней челюсти, либо уменьшают её [5] [6] [7] . При этом развивается соответствующая симптоматика в виде нарушений прикуса, функционального нарушения дыхания и эстетических диспропорций строения лица [8] [9] [10] .
Классификация и стадии развития микрогнатии
Разделение дисгнатий на классы связано с видом нарушения прикуса. В зависимости от вида смыкания зубных рядов по молярам и клыкам американский стоматолог Э. Энгль разделил все аномалии прикуса на три класса:
- I класс — нейтральный, правильный прикус — нормальное соотношение зубных рядов относительно друг друга.
- II класс — дистальный, задний прикус: т. е. смещение нижнего зубного ряда кзади:
- I подкласс — передние зубы верхней челюсти наклонены в сторону губ, т. е. вперёд;
- II подкласс — передние зубы верхней челюсти наклонены в сторону нёба, т. е. внутрь.
- III класс — мезиальный, передний прикус — смещение нижнего зубного ряда вперёд [14] .
Микрогнатия будет характерна для двух классов аномалий прикуса по Энглю:
- дисгнатии II класса — сопровождается недоразвитием нижней челюсти;
- дисгнатии III класса — сопровождается недоразвитием верхней челюсти.
Т. о., дисгнатия II класса всегда ассоциируется с дистальным прикусом, а дисгнатия III класса — с мезиальным.
Отдельно стоит сказать о врождённой микрогнатии нижней челюсти, связанной с гемифациальной микросомией — ростом одной стороны лица, который приводит к асимметрии или недоразвитию некоторых органов: ушей, рта и нижней челюсти и др. Её классифицируют по степеням тяжести:
- I степень — уменьшение ветви нижней челюсти и ямки височной кости с сохранением других структур.
- II степень — деформация и недоразвитие других структур нижней челюсти, помимо вышеописанных, с нарушением или сохранением работы височно-нижнечелюстного сустава.
- III степень — несформированность височно-нижнечелюстного сустава, отсутствие его структур.
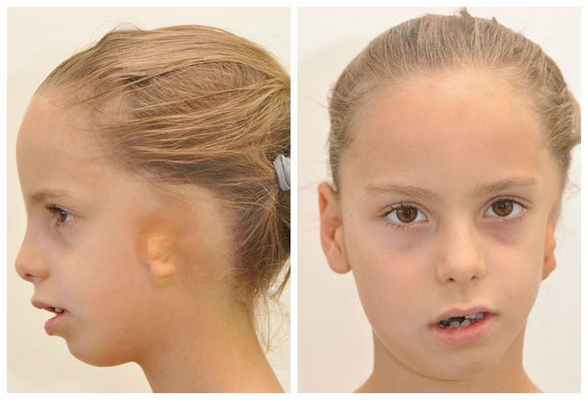
Осложнения микрогнатии
В большинстве случаев недоразвитие челюсти даёт о себе знать ещё в детстве, поэтому приступать к его лечению можно и нужно сразу, не затягивая. Иначе длительное существование подобных скелетных нарушений обязательно скажется на функционировании и здоровье челюстно-лицевой системы. Откладывание лечения чревато развитием заболеваний пародонта, более частым образованием кариеса, возникновением симптомов нарушения работы височно-нижнечелюстного сустава: перегрузок, болей, щелчков и хруста при открывании рта.
Среди возможных осложнений также можно выделить следующие:
- заболевания желудочно-кишечного тракта — вызваны некачественным пережёвыванием продуктов; — связано с недополучением части питательных элементов из-за проблем с пережёвыванием пищи;
- нарушение дыхательной функции и развитие заболеваний органов дыхания;
- нарушение мозгового кровоснабжения — связано с нарушением дыхания, которое приводит к недостаточному насыщению сосудов головного мозга кислородом;
- дефекты дикции.
Кроме того, неестественный размер челюсти становится причиной асимметрии овала лица, что неизбежно отражается на привлекательности пациента и его уверенности в себе. На этой почке нарастает психологический дискомфорт человека, его тревожность и склонность к депрессии. Также эстетические нарушения влияют на восприятие человека в социуме, отражаясь на его работе и личной жизни.
В целом присутствие подобных аномалий негативно сказывается на качестве жизни, заметно снижая её [15] . Чтобы сохранить здоровье и предотвратить развитие последствий микрогнатии следует обратиться за помощью к специалистам.
Диагностика микрогнатии
В целях диагностики микрогнатии в обязательном порядке оцениваются:
- вид смыкания зубных рядов — определяют с помощью диагностических гипсовых моделей (при необходимости их устанавливают в артикулятор);
- состояние ротовой полости — клинический осмотр;
- фотометрические характеристики лица, улыбки, головы и верхней половины тела — нужны для анализа контуров мягких тканей, смыкания зубных рядов, работы лицевых и жевательных мышц в динамике.
Основным современным видом лабораторной диагностики микрогнатии является компьютерная рентгеновская томография. В идеале она выполняется на конусно-лучевых томографах, используемых в челюстно-лицевой диагностике. У них довольно широкая область сканирования — не менее 17-23 см. Это позволяет визуализировать весь лицевой череп [16] . После обработки таких томограмм с использованием специальных программ (например Dolphin) можно досконально изучить полученные снимки, выявить вид деформации и создать виртуальный лечебный план.

В некоторых случаях дополнительно требуется выполнить МРТ структур височно-нижнечелюстных суставов [17] . При подозрении на дыхательные нарушения пациентам проводят полисомнографические исследования — диагностику нарушений сна путём установки специальных датчиков. Вместе все эти данные влияют на построение лечебного плана, позволяя получить необходимый результат терапии.
Лечение микрогнатии
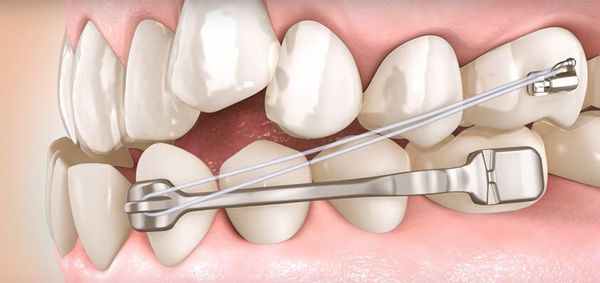
Лечение большинства невыраженных скелетных нарушений с сопутствующими нарушениями прикуса проводят стомато логи-ортодонты. Для устранения возникших проблем они применяют различные ортодонтические системы:
- съёмная аппаратура — элайнеры, ортодонтические пластинки, лицевые маски [18] ;
- несъёмная аппаратура — брекеты [19] .
При выраженных скелетных нарушениях с сопутствующими значимыми функциональными и эстетическими нарушениями применяется комбинированное ортодонтическо-хирургическое лечение. Оно направлено на устранение эстетических и функциональных нарушений: исправление прикуса, изменение формы и позиции костных структур лица.
От ортодонтов зависит качественное смыкание зубных рядов после выполнения челюстно-лицевой операции и окончательный результат комплексного лечения. Хирурги же перемещают челюсти (одну или обе) и изменяют их форму в случае необходимости. После перемещения челю сти они придают ей правильное положение и закрепляют специальными фиксирующими элементами. Для скрепления используют специальные пластины из медицинского титана. В некоторых случаях для небольшой коррекции контуров лица (подбородка, скул, углов нижней челюсти) используются имплантаты.
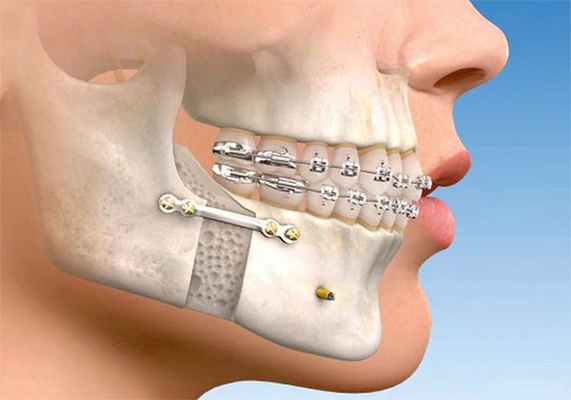
Такой вид хирургии требует детального планирования. Операция проводится в условиях наркоза из внутриротового доступа [20] . После неё пациент наблюдается в стационаре в течение 1-2 дней [21] . При полном сращении челюстных костей (примерно через 14 недель) ортодонт заканчивает выравнивание зубов с применением брекет-системы.
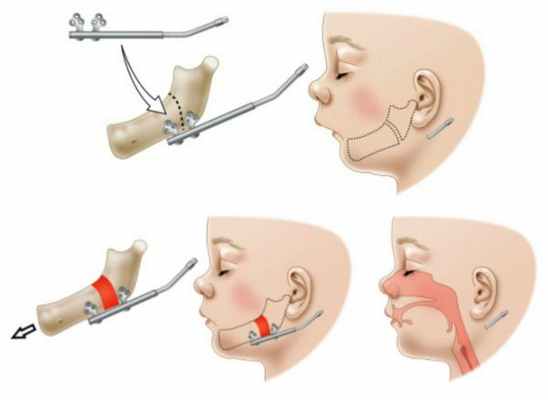
В случае микрогнатии с функциональными расстройствами дыхания в младенческом возрасте показан дистракционный остеогенез нижней челюсти. Данная операция заключается в формировании нижней челюсти путём медленного растяжения кости.
Прогноз. Профилактика
Единожды исправив функционально-эстетические нарушения челюстных костей и восстановив гармонию лица, пациенты забывают о ранее существующих проблемах навсегда. При условии точного предоперационного планирования хирургическое вмешательство приводит к стойкому положительному результату. По завершении периода реабилитации и окончании ортодонтического лечения результат не требует дополнительной коррекции на протяжении жизни.
Эстетическая сторона вопроса чаще всего стоит на первом месте для пациентов, готовящихся избавиться от микрогнатия [22] . Поэтому челюстно-лицевые хирурги совместно с ортодонтами всегда работают над получением максимально эстетических и функциональных результатов. Человек начинает чувствовать уверенность в себе и своей внешности, получает новые возможности в жизни и карьере, качество их жизни заметно улучшается.
Читайте также: