Синдром Робертса у плода. Диагностика и прогноз при синдроме Робертса
Добавил пользователь Валентин П. Обновлено: 28.01.2026
Робертса синдром (J. Roberts, 1919), или тетрафокомелия с расщелиной губы и нёба, - наследственное заболевание с множественными пороками развития (редукционными пороками конечностей, расщелинами верхней губы и нёба, пороками развития глаз и других органов), часто несовместимыми с жизнью. Тип наследования - аутосомно-рецессивный.
Клиническая симптоматика
Тяжёлая форма болезни проявляется с рождения микроцефалией с выраженным гипертелоризмом и птозом, экзофтальмом и голубыми склерами, часто - с врожденными помутнениями роговиц (склеророговица), с врождённой катарактой и глаукомой, а также с гипоплазией крыльев носа и коротким фильтром, двусторонней расщелиной верхней губы и нёба, с треугольной формой рта, деформированными ушными раковинами, с капиллярными гемангиомами на лице и редкими серебристо-белыми волосами.
Тетрафокомелия - характерная для этого синдрома патология; верхние конечности обычно затронуты патологическим процессом больше, чем нижние; тяжесть этих нарушений варьирует от редукции конечностей до фокомелии (чаще отсутствуют кости предплечья и голени); примерно у половины этих больных отсутствуют проксимальные отделы конечностей (длина конечностей зависит от степени их гипоплазии); нередко выявляют верхнюю олигодактилию, синдактилию, анкилозы, флексорные контрактуры локтевых и коленных суставов, косолапость, гипоплазию ногтей и изменение дерматоглифики, а также черепно- и спинномозговые грыжи, врождённые пороки сердца, аномалии почек, гипоплазию полового члена или клитора, крипторхизм, двурогую матку.
Прогноз в отношении жизни: многие дети с этим синдромом уже рождаются мёртвыми или погибают вскоре после рождения в связи с врождёнными пороками, не совместимыми с жизнью.
Синдром Пьера Робена ( Аномалия Робена , Последовательность Робена )
Синдром Пьера Робена - это генетически обусловленная аномалия, характеризующаяся гипоплазией нижней челюсти, небной расщелиной и глоссоптозом. Комбинация пороков ЧЛО вызывает затруднения при вскармливании, обструктивное апноэ, диффузный цианоз, риск аспирации пищи и асфиксии. Патология диагностируется по данным рентгенографии и КТ челюсти, фиброларингоскопии, полисомнографии, генодиагностики. Консервативная тактика включает позиционную терапию, СРАР, ортодонтическое лечение. Методы хирургической коррекции порока представлены глоссопексией, пластикой неба, компрессионно-дистракционным остеосинтезом.
МКБ-10



Общие сведения
Синдром (последовательность, аномалия) Робена включает триаду признаков: микрогнатию, глоссоптоз, расщепление твердого и мягкого неба. Врожденная анатомическая деформация описана в 1923 г. французским зубным врачом Пьером Робеном. По разным оценкам, дети с данной аномалией рождаются с популяционной частотой 1:8500-30000, независимо от пола. Ведущей проблемой младенцев с синдромом Робена является обструкция верхних дыхательных путей и критическое нарушение дыхания вследствие недоразвития нижней челюсти и смещения мягких тканей вглубь полости рта.

Причины
Последовательность Пьера Робена может встречаться как изолированный синдром (20-40%) или как часть других генетических патологий. В первом случае челюстно-лицевая аномалия формируется под влиянием неблагоприятных условий внутриутробного развития, в числе которых может быть:
- механическое сдавление плода в полости матки, ограничивающее рост челюсти (рубцами, амниотическими тяжами, опухолями или другим плодом при многоплодной беременности);
- внутриутробные инфекции;
- маловодие;
- дефицит фолиевой кислоты в организме беременной;
- тератогенные воздействия на плод, вызванные вредными экологическими факторами, радиацией, химическими веществами, приемом ЛС.
Кроме этого, аномалия Пьера Робена встречается в структуре генетических синдромов с множественными пороками развития. Всего насчитывается порядка 300 таких заболеваний, среди которых синдромы Стиклера, Хангарта, Эдвардса, кампомелическая дисплазия и др. Более чем в 30% случаев синдром Пьера Робена сочетается с ВПС, скелетными аномалиями, дефектами развития глаз и ушных раковин.
Спорадические случаи возникают в результате генетических мутаций de novo. Синдромальная последовательность наследуется по тому же принципу, что и патология, с которой она ассоциирована (сообщается об аутосомно-рецессивном, аутосомно-доминантном, Х-сцепленном варианте). Согласно исследованиям, генетические мутации затрагивают области хромосом 2 (2q24.1-33.3), 4 (4q32-qter), 11 (11q21-q23.1) и 17 (17q21-q24.3).
Патогенез
Синдром Пьера Робена принято называть «последовательностью», поскольку его признаки представляют собой цепочку взаимовлияющих друг на друга событий. Нарушение развития нижней челюсти у плода происходит в период между 7-й и 11-й неделями гестации под воздействием генетических мутаций или экзогенных факторов. Гипоплазия челюсти способствует неправильному положению языка между небными пластинками, которое препятствует их своевременному закрытию и вызывает образование расщелины.
Нижняя ретрогнатия и маленький объем ротовой полости создают условия для смещения языка в сторону задней стенки глотки и возникновения глоссоптоза. Оттянутый назад язык вызывает у новорожденного серьезные затруднения дыхания, вследствие чего часто возникает инспираторная обструкция ВДП.
Классификация
В зависимости от выраженности нарушения глотания и дыхания выделяют три степени тяжести синдрома Пьера Робена:
- легкая - дыхание не нарушено, имеются небольшие затруднения при кормлении младенца, которые устраняются консервативными методами в домашних условиях;
- средняя - трудности дыхания и кормления выражены умеренно, ребенку требуется стационарная помощь, зондовое питание;
- тяжелая - выраженные затруднения дыхания и самостоятельного питания, требующие постановки трахеостомы и желудочного зонда.
Симптомы
Новорожденные с синдромом Пьера Робена имеют характерное «птичье» лицо, обусловленное гипоплазией нижней челюсти (микрогнатия) и ее смещением назад (ретрогнатия). Объем полости рта полости уменьшен, язык оттянут вглубь (глоссоптоз). Как правило, у пациентов обнаруживается U-образная субмукозная расщелина мягкого неба, также встречается готическое небо.
Смещение языка кзади вызывает проблемы с сосанием в раннем возрасте, из-за чего у ребенка отмечается задержка роста и набора веса, отставание в моторном развитии. Тяжесть дыхательных нарушений варьируется от храпа до опасных эпизодов обструктивного апноэ. В легких случаях дыхательные расстройства возникают только в положении на спине и при беспокойстве ребенка, проходят самостоятельно. В более серьезных ситуациях приступы остановки дыхания длительные (до 20 сек. и более), приводящие к развитию гипоксии и гиперкапнии.
Тяжелые эпизоды апноэ сопровождаются акроцианозом или диффузным цианозом. Постоянная задействованность вспомогательных дыхательных мышц способствует формированию воронкообразной деформации грудной клетки.
Синдромальная последовательность Пьера Робена, как правило, сочетается с врожденными аномалиями других органов: тугоухостью и недоразвитием слухового аппарата, патологией зрительного анализатора (близорукость, катаракта), скелетными деформациями (полидактилия, врожденные ампутации), сердечными пороками. Примерно у половины пациентов обнаруживаются дефекты ЦНС: микроцефалия, гидроцефалия, эпилепсия, задержка психоречевого развития, олигофрения.
Осложнения
Практически все дети с синдромом Пьера Робена страдают речевыми дефектами (открытая ринолалия), нарушением прикуса, рецидивирующими ушными инфекциями. На фоне пролонгированных приступов обструктивного апноэ постепенно формируется гипоксическая энцефалопатия, легочное сердце. В некоторых случаях ребенок может погибнуть от асфиксии. Дисфагические расстройства могут повлечь за собой аспирацию слюны, пищи, желудочного содержимого при срыгивании, что чревато развитием аспирационной пневмонии. Смертность детей составляет порядка 16%, а при наличии сочетанных пороков достигает 41%.
Диагностика
В настоящее время синдром Пьера Робена диагностируется пренатально с помощью скринингового УЗИ в 11-12 недель беременности. После рождения диагноз подтверждается данными клинической картины и инструментальных исследований. Необходимая диагностика:
- Рентгеновские методы. Для первичной оценки анатомии челюстно-лицевой зоны выполняется рентгенография нижней челюсти, гортани и трахеи. В рамках предоперационного обследования обязательно проведение КТ челюстей, лицевого черепа, мягких тканей шеи. Рентгеновская визуализация помогает определить степень микро- и ретрогнатии, обструкции дыхательных путей, рассчитать параметры дистракции.
- Эндоскопия гортани. С помощью фиброларингоскопии уточняется состояние носоглотки, гортани и начальных отделов трахеи. Оценивается уровень и степень обструкции ВДП, обнаруживаются другие аномалии ЛОР-органов.
- Полисомнография. Считается «золотым стандартом» определения тяжести синдрома обструктивного апноэ. С ее помощью выясняется частота и длительность эпизодов остановки дыхания, подбирается оптимальная терапия.
- Другие методы. Регулярно проводится исследование газового состава крови, пульсоксиметрия. Перед планируемой операцией выполняется фотофиксация внешнего вида ребенка в фас и профиль, регистрация прикуса. При синдромальных формах аномалии Робена показана генодиагностика.
Лечение синдрома Пьера Робена
Лечебная тактика при аномалии Робена зависит от выраженности респираторных и дисфагических расстройств. Она может заключаться в проведении позиционной терапии, вспомогательной терапии, хирургического лечения. На различных этапах жизни ребенку требуется сопровождение врача-отоларинголога, челюстно-лицевого хирурга, ортодонта, логопеда.
Консервативное лечение
При легкой степени дыхательной обструкции рекомендуется постуральная терапия. Ребенка выкладывают на живот или на бок с приподнятой или зафиксированной головой, обеспечивающей выдвижение вперед языка и нижней челюсти. Персистирующая обструкция может купироваться с помощью назофарингеального воздуховода. Одним из методов с подтвержденной эффективностью является СРАР-терапия, позволяющая осуществлять неинвазивную масочную вентиляцию под постоянным положительным давлением.
При невозможности самостоятельного питания кормление ребенка производится через назогастральный зонд. Тяжелые степени синдрома Пьера Робена требуют установки трахеостомы или интубации трахеи с подключением ребенка к аппарату ИВЛ.
Консервативные методы позволяют купировать симптомы, но ни один из них не устраняет имеющиеся врожденные аномалии. Поэтому такое лечение должно проводиться очень длительно, до тех пор, пока размеры дыхательных путей ребенка не позволят ему дышать самостоятельно без риска обструкции. После нормализации жизненных функций проводится ортодонтическое лечение, логопедическая коррекция.
Хирургическое лечение
Устранение небной расщелины проводится поэтапно: велофарингопластика выполняется в 6-8 мес., уранопластика - в 12-18 мес. В качестве временной меры при подготовке к радикальному лечению может осуществляться глоссопексия - фиксация языка к нижней губе, альвеолярному отростку. Ранее применявшиеся методы вытяжения нижней челюсти, транспозиции жевательных мышц не являются достаточно эффективными.
Радикальным хирургическим вмешательством, позволяющим удлинить нижнюю челюсть, является компрессионно-дистракционный остеосинтез. Операция заключается в проведении двусторонней остеотомии нижней челюсти с фиксацией на костных фрагментах специального аппарата. Активация устройства обеспечивает постепенное перемещение нижней челюсти вместе с комплексом мягких тканей дна полости рта кпереди. Процесс дистракции занимает около 3-х месяцев, в результате чего становится возможным сначала самостоятельное дыхание, а затем - питание.
Прогноз и профилактика
Изолированный синдром Робена имеет более благоприятный прогноз, чем синдромальные формы, ассоциированные с множественными пороками и умственной отсталостью. Также существенное значение имеет своевременность и радикальность лечения. Вместе с тем, на данный момент смертность от асфиксии, других осложнений и сопутствующих аномалий остается достаточно высокой.
Для снижения риска рождения ребенка с синдром Пьера Робена рекомендуется не допускать воздействия тератогенов, инфицирования, дефицита микронутриентов в период гестации. Беременным необходимо вовремя проходить генетические и ультразвуковые пренатальные скрининги.
1. Синдром Пьера Робена у детей/ Кириллова Л.Г., Ткачук Л.И., Шевченко А.А., Силаева Л.Ю., Лисица В.В., Мироняк Л.А.// Международный неврологический журнал. - 2010. - №3 (33).
3. Хирургическое лечение новорожденных и грудных детей с синдромом Пьера Робена/ Дубин С.А., Комелягин Д.Ю., Злыгарева Н.В., Строгонов И.А.,Р огинский В.В., Полуэктов М.Г.// Российский вестник детской хирургии, анестезиологии и реаниматологии. - 2011.
4. Клинический случай синдрома Пьера Робена у новорожденного ребенка/ Вахитова Л.Ф., Фазлеева Л.К., Булгакова Л.Г., Варламова О.В.// Практическая медицина. - 2013.
Синдром Жубер
Синдром Жубер - редкое генетически гетерогенное наследственное заболевание, характеризующееся нарушением формирования мозжечка и структур мозгового ствола с развитием соответствующей неврологической симптоматики. Симптомы данной патологии проявляют значительную вариабельность по своей выраженности, наиболее часто наблюдаются расстройства дыхания, глазодвигательные нарушения и мышечная слабость, возможны нарушения слуха и отставание в интеллектуальном развитии. Диагностика синдрома Жубер производится на основании неврологического осмотра больного, магнитно-резонансной томографии головного мозга, молекулярно-генетических исследований. Специфического лечения на сегодняшний момент не существует, применяют поддерживающие и симптоматические мероприятия.

Синдром Жубер (Жуберт) - достаточно редкая генетическая патология, которая характеризуется нарушением эмбрионального развития важных структур головного мозга, что приводит к различным неврологическим проблемам. Впервые данное заболевание было описано канадским педиатром Мари Жуберт в 1969 году. Она выявила у четырех детей, чьи родители состояли между собой в кровном родстве, нарушения дыхания, слуха, мышечную слабость и признаки умственной отсталости. С 1977 года подобные нарушения выделили в отдельную нозологическую единицу под названием «синдром Жубер». В дальнейшем врачи-генетики смогли определить значительную генетическую гетерогенность заболевания - на сегодняшний момент известно не менее 20 генов, мутации которых смогли связать с этой патологией. При этом почти в половине случаев синдрома Жубер его молекулярно-генетические механизмы остаются неизвестными. По последним данным, встречаемость этого заболевания составляет примерно 1 случай на 1 млн. новорожденных, механизм передачи аутосомно-рецессивный, мальчики и девочки поражаются с одинаковой частотой. Каких-либо национальных, расовых или региональных особенностей в распределении синдрома Жубер не выявлено.

Причины и классификация синдрома Жубер
По мере изучения синдрома Жубер обнаруживается все больше генов, мутации которых способны приводить к развитию этого заболевания. На сегодняшний день таких генов выявлено почти два десятка, при этом примерно в половине клинических случаев синдрома Жубер определить генетические нарушения не удается, что говорит о роли других, еще не изученных дефектов. Мутации генов, приводящие к этому заболеванию, возникают на различных хромосомах, но всегда наследуются по аутосомно-рецессивному механизму. Также все эти гены объединяет участие в эмбриональном развитии структур головного мозга, именно это обстоятельство служит причиной появления характерной симптоматики синдрома Жубер.
Установлено, что мутации одних генов приводят к развитию заболевания несколько чаще, чем другие. Одним из них является ген AHI1, который располагается на 6-й хромосоме. Согласно данным медицинской статистики, дефекты этого гена становятся причиной 10-12% всех случаев синдрома Жубер. Ген AHI1 кодирует специфический белок, принимающий активное участие в формировании некоторых элементов ствола мозга и сетчатки глаза. Другой распространенной причиной синдрома Жубер считается дефект гена CEP290, локализованного на 12-й хромосоме. Он кодирует последовательность белка, принимающего участие в формировании клеточных центросом и ресничек в различных органах организма (головной мозг, сетчатка глаза, сердце, легкие, почки). Мутации гена CEP290 выявляются примерно у 10% больных синдромом Жубер.
Кроме того, к синдрому Жубер могут приводить мутации таких генов, как TCTN1 и TCTN2 (12-я хромосома), TMEM138 и TMEM216 (11-я хромосома), TMEM237, TTC21B и NPHP1 (2-я хромосома), ARL13B (5-я хромосома) и целый ряд других. Они встречаются с частотой в несколько процентов от всех случаев заболевания, для многих относительная распространенность неизвестна. Те гены, функции которых удалось установить, также контролируют развитие ресничек, центриолей или цитоскелета, что позволяет считать синдром Жубер проявлением нарушений формирования именно этих структур. Мутации всех вышеперечисленных генов наследуются по аутосомно-рецессивному механизму, однако в последние годы появились указания на возможность сцепленной с полом передачи. Предполагают, что такая форма синдрома Жубер обусловлена мутацией гена OFD1, локализованного на Х-хромосоме.
Симптомы синдрома Жубер
Фенотипические проявления синдрома Жубер в целом сходны при различных генетических разновидностях заболевания. Вместе с тем, имеются незначительные различия. Выраженность симптоматики может значительно различаться даже в пределах одной семьи. В настоящее время достоверно неизвестны причины того, почему тяжесть течения синдрома Жубер отличается у разных больных. В подавляющем большинстве случаев заподозрить наличие заболевания можно в первые дни жизни ребенка - выявляется мышечная гипотония, аномальные движения глаз, часто определяется колобома (дефект оболочек глаза). Характерным признаком синдрома Жубер являются нарушения дыхания - нестабильность ритма (тахипноэ, брадипноэ), возможна остановка дыхания во время сна (ночное апноэ).
По мере роста ребенка отмечается незначительное прогрессирование заболевания - мышечная гипотония перетекает в мозжечковую атаксию, наблюдается отставание в моторном и интеллектуальном развитии. При этом спектр нарушений интеллекта при синдроме Жубер колеблется в очень широких пределах - от нормы до глубокой умственной отсталости. Возможны нарушения слуха (вплоть до нейросенсорной глухоты) и зрения (обусловленные как глазодвигательными аномалиями, так и дистрофией сетчатки). Примерно у половины больных синдромом Жубер развиваются различные аномалии внутренних органов - фиброз печени, поликистоз почек, врожденные пороки сердца. Реже возникает энцефалоцеле (через большое затылочное отверстие или дефекты свода черепа), гидроцефалия, гамартомы полости рта.
Из-за значительного диапазона выраженности проявлений синдрома Жубер риски летального исхода и различных осложнений достаточно неопределенные. В самых тяжелых случаях возможна смерть больных в младенческом возрасте из-за дыхательных и неврологических нарушений. При менее тяжелом течении синдрома Жубер пациенты могут доживать до взрослого и даже преклонного возраста, при этом они, как правило, лишены возможности ходить из-за мозжечковой атаксии. Основные риски в подобных случаях создают поражения и аномалии развития внутренних органов - сердца, почек, печени.
Диагностика и лечение синдрома Жубер
Для определения синдрома Жубер применяют следующие диагностические методики: неврологический осмотр больных, магнитно-резонансную томографию головного мозга, дополнительные исследования глаз, слуха и работы внутренних органов. Молекулярно-генетическая диагностика в большинстве современных лабораторий возможна в отношении четырех наиболее распространенных типов заболевания - обусловленных мутациями генов AHI1, CEP290, CC2D2A и TMEM67. При осмотре больных синдромом Жубер определяется мышечная слабость, признаки мозжечковой атаксии и нарушений координации, основные сухожильные рефлексы резко снижены. Практически всегда обнаруживается отставание в моторном развитии, в ряде случаев - различная степень умственной отсталости.
Самым типичным диагностическим признаком синдрома Жубер является наличие так называемого «симптома молярного зуба» - характерные изменения на МРТ головного мозга, внешне похожие на разрез зуба. Это проявление говорит о наличии нарушений формирования стволовых элементов мозга. Также на магнитно-резонансной томографии часто определяется недоразвитие червя мозжечка, гипоплазия мозолистого тела, гидроцефалия, расширение желудочков, энцефалоцеле и другие аномалии развития головного мозга. У взрослых больных синдромом Жубер нередко выявляются признаки поражения внутренних органов - поликистоз почек, фиброз печени, нарушения сердечного ритма. При осмотре у офтальмолога часто обнаруживаются непроизвольные аномальные движения глаз (нистагм), колобома, дистрофия и дегенерация сетчатки.
При помощи методов современной генетики возможна молекулярно-генетическая диагностика синдрома Жубер, который вызывается мутациями генов AHI1, CEP290, CC2D2A и TMEM67. В общей сложности дефекты этих генов обуславливают порядка 40% всех случаев заболевания. По этой причине отрицательный результат генетических анализов не является поводом для гарантированного исключения синдрома Жубер. Вспомогательную роль в определении патологии играет изучение наследственного анамнеза больного с целью подтверждения аутосомно-рецессивной передачи. При помощи прямого автоматического секвенирования можно выявлять носительство патологической формы гена у родственников больного или при отягощенной по этому состоянию наследственности.
Специфического лечения синдрома Жубер не существует, медицинская помощь при этом заболевании сводится к паллиативным и симптоматическим мероприятиям. Для ослабления неврологических симптомов применяют ноотропные средства - их регулярный прием, начатый с раннего возраста, может значительно улучшить прогноз в отношении интеллектуального развития больного. Также при синдроме Жубер используют различные методы физиотерапии, специальные упражнения для улучшения координации движений и уменьшения проявлений атаксии. В раннем возрасте часто необходим контроль дыхания больного во избежание потенциально опасного апноэ.
Прогноз и профилактика синдрома Жубер
Прогноз синдрома Жубер часто неопределенный, поскольку находится в зависимости от выраженности симптомов и тяжести клинического течения заболевания. В самых тяжелых случаях возможен летальный исход еще в раннем детстве из-за нарушений дыхания и неврологических патологий. В большинстве случаев больные доживают до взрослого и даже пожилого возраста, хотя совокупность атаксии, патологий внутренних органов, нарушений зрения и слуха часто приводит к глубокой инвалидизации. Умственное развитие может быть сохранено, но встречаются и различные степени умственной отсталости. Профилактика синдрома Жубер возможна только в качестве пренатальной диагностики заболевания и определения носительства патологической формы гена у лиц с отягощенной наследственностью.
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма
Эталон новых стандартов! Беспрецедентная четкость, разрешение, сверхбыстрая обработка данных, а также исчерпывающий набор современных ультразвуковых технологий для решения самых сложных задач диагностики.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) - это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза-Франческетти, синдром Франческетти-Цвалена-Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер - имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза-Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
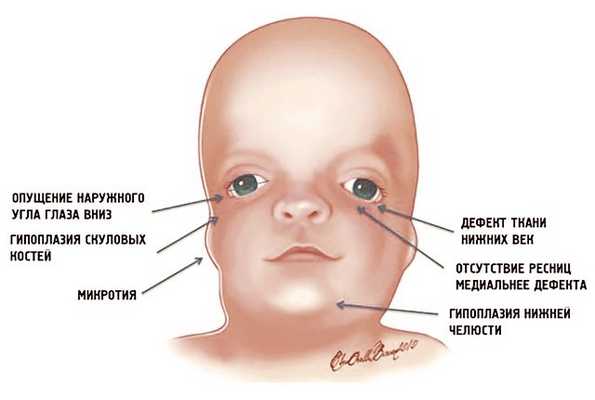
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса 1, также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости 5. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев - это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 - q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4-6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип - мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши - все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности - редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким - 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12-13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2-4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
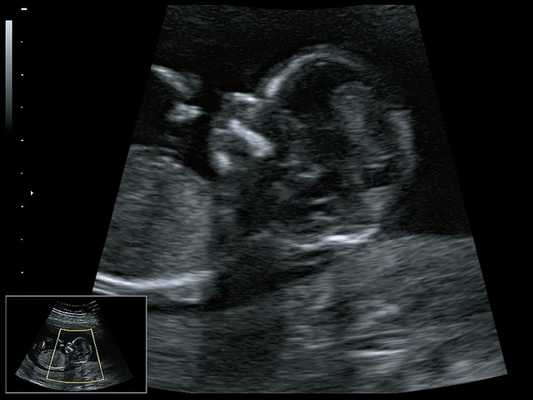
Рис. 2. Микрогнатия - сагиттальный скан в 2D, беременность 13 нед.
Синдром Эдвардса: короткая, но полная любви жизнь

Еще есть время, чтобы быть рядом — и любить здесь и сейчас
Информация про синдром Эдвардса на всех медицинских ресурсах неутешительная: прогноз для этого заболевания весьма неблагоприятный. Но вопреки заложенному сценарию развития заболевания многие дети при хорошем уходе и паллиативной помощи живут довольно долго. Лекарство от заболевания одно — родительская любовь.
«Такие множественные пороки — это, скорее всего, генетика»
Саше Глаголевой поставили синдром Эдвардса вскоре после рождения. Сейчас Саше 8 лет, хотя во всех медицинских справочниках написано, что девочки с таким заболеванием живут в среднем около 10 месяцев, а мальчики от двух до трех месяцев, и вообще около 90% детей с синдромом Эдвардса умирают в течение первого года жизни.
Саша родилась со множественными пороками развития, и врачи в реанимации сразу предположили генетическое заболевание.
Родители не могли поверить, ведь и сами они были здоровы, и у них уже был совершенно здоровый ребенок, но врачи объяснили, что дети с генетическими проблемами иногда появляются из-за спонтанной мутация в генах или хромосомах, а не только из-за наследственности.
На третий день генетический тест подтвердил предположение врачей — синдром Эдвардса.
Что такое синдром Эдвардса
В описаниях заболевания сказано, что в 60% случаях из-за пороков, несовместимых с жизнью, дети с синдромом Эдвардса погибают внутриутробно. В группе трисомий только в трех случаях — при синдроме Эдвардса, синдроме Дауна (трисомия 21-ой хромосомы) и синдроме Патау (трисомия 13-ой хромосомы) возможно рождение живого ребенка и его дальнейшее, хоть и осложненное, развитие . При других вариантах добавочных хромосом патология несовместима с жизнью.
Как и другие заболевания с хромосомными мутациями, синдром Эдвардса неизлечим. В настоящее время лечение состоит, в основном, из паллиативной помощи.
Как диагностируют синдром Эдвардса
Заподозрить наличие синдрома Эдвардса можно на УЗИ плода и допплерографии по косвенным признакам: недоразвитию одной из пупочных артерий, многоводию, уменьшению размеров плаценты. Но ультразвуковое исследование может показать только характерные признаки патологии, оно не может подтвердить, что есть именно этот синдром.

Эпоха Александры Евгений Глаголев, отец тяжелобольной девочки рассказывает о том, как изменилась жизнь его семьи, почему он решил сменить профессию и зачем родителям время от времени выезжать куда-то вдвоем
Вообще, для диагностики заболевания большое значение имеет совокупность факторов: результаты лабораторного скрининга, данные УЗИ, срок беременности и возраст матери. Если есть сразу несколько симптомов необходимо провести дополнительные инвазивные исследования — биопсию ворсин хориона ( анализ клеток эмбриональной ткани) , амниоцентез (взятие на анализ околоплодных вод) или кордоцентез (взятие анализа крови из пуповины). Полученный в ходе процедур материал плода отправляется на кариотипирование — анализ на выявление нарушений хромосомного набора.
У новорожденного ребенка для определения точной причины врожденных дефектов тоже должно быть проведено хромосомное исследование с помощью анализа крови.
«Моя дочь растет, развивается, она личность и просто ребенок»
После того, как ее дочери поставили синдром Эдвардса, Екатерина оказалась буквально в информационном вакууме, и все, что она прочитала о болезни, было очень пессимистичным относительно прогноза жизни таких детей. Она начала общаться с другими родителями детей с трисомией и решила объединить их в сообщество , в котором сейчас более 40 семей.
По ее мнению, данные в справочниках по генетике по этому заболеванию устарели — они написаны 20-30 лет назад, а ведь за это время медицина шагнула далеко вперед, и выхаживание детей с синдромом Эдвардса стало возможным. Она понимает, что вылечить синдром нереально, но уверена, что облегчить состояние ребенка можно.

Каждая секунда имеет значение и смысл История Ани и Славы Черепановых. Родителей, потерявших двоих детей
По мнению Екатерины, это свидетельствует о том, что несмотря на тяжесть состояния и высокую смертность, синдром Эдвардса некорректно рассматривать как летальное и несовместимое с жизнью заболевание: «Моя дочь растет, развивается, она личность и просто ребенок, хоть и не нормотипичный, но со своим характером и потребностями».
Программа перинатальной паллиативной помощи
Если диагноз трисомии поставлен до рождения ребенка, у родителей есть выбор — прервать беременность или родить неизлечимо больного ребенка. Семьи, которые считают прерывание беременности недопустимым, поддерживает Д етский хоспис «Дом с маяком». Специалисты хосписа находятся рядом с родителями при родах и потом все время, пока паллиативный ребенок дома, сопровождают семью.
Хоспис обеспечивает такие семьи необходимым оборудованием, расходными материалами, лекарствами, специальным питанием, обучает родителей навыкам ухода за малышом. Кроме того, семью курируют врачи, психологи, социальные работники. Еще есть программа социальной передышки — можно оставить ребенка в стационаре на две недели в году, чтобы родители могли отдохнуть.
Первым ребенком, рожденным под опекой «Дома с маяком», была Маруся. Ее мама написала , что ей было очень сложно получить хоть какую-то информацию о том, какими рождаются дети с синдромом Эдвардса: «Я пересмотрела и перечитала терабайты историй западных семей с такими детьми, и они были очень светлыми, несмотря на весь мрак ситуации, я знала, что дети с синдромом Эдвардса бывают весьма крепкими, что совсем необязательно там будет страшный болевой синдром, судороги и асфиксия, что у них может быть короткая, но полная любви и заботы жизнь, однако как реализовать это в наших условиях, я не понимала. Результаты моих обследований говорили о том, что у Маруси были высокие шансы на благополучное рождение и хороший светлый промежуток».
Женщина обратилась в Детский хоспис. Потом она познакомилась с семьей Глаголевых, а Вероника Машкова рассказала ей, каким был ее сын Коля.
«Три месяца мы жили почти обычной жизнью. Маруся была очень славной, почти обычной девочкой. Мы начали гулять. Я строила планы лечения и реабилитации. Последний месяц был сложнее. За три дня произошло резкое ухудшение. Мне до сих пор сложно дается осознание того, что же все-таки произошло. Я не хотела верить, что наше время вышло, и здесь очень четко сработал врач «Дома с маяком»».

Светлая печаль, как сама жизнь
Сын Вероники Машковой прожил три месяца. Коля тоже родился с синдромом Эдвардса. Вероника часто делится опытом переживания потери на встречах с родителями, потерявшими детей .
«Каждый раз возникает какое-то движение. Сначала приходит родитель, который вообще не может говорить, просто давится слезами, которые даже не текут. А со временем это живой, разговорчивый, общительный человек — да, плачущий, но это добрые слезы. Да, это потеря, это боль, ее стоит выплакать».
Вероника рассказывает, что сначала на этих встречах обычно просто молча слушала всех, кто пришел. Могла сказать что-то в конце, что отозвалось. Но часто поражалась: «Я не раз видела это чудо: родители приходили раздавленными, а поднимались распрямившимися и окрыленными, супруги приходили порознь, а уходили за руку…
Да, у нас тоже бывает печаль, но она светлая, как сама жизнь. Это ведь радость, что наши дети у нас были, и мы смогли это время вместе с ними прожить. А меня очень сильно поддерживало то, что все было не напрасно, что жизнь Коли и его история нужны людям».
У меня еще есть время, чтобы быть рядом
«Нам очень много помогал и помогает фонд «Вера» , — рассказывает Екатерина Глаголева, — На момент, когда мы выписались первый раз из больницы, мы не знали ничего — что и как с Сашей делать. Фонд нас очень быстро «подхватил» — приехала медсестра, привезли кислородный концентратор, отсос, расходные материалы, нас научили, куда что вставлять. И стало не так страшно. Чувствовалась настоящая поддержка. Особенно актуальная на фоне округляющихся глаз врачей, которые наблюдают Сашу в районной поликлинике. Хотя у нас очень хороший участковый педиатр, но она сама признает, что таких детишек на участке больше нет, и она не знает, что делать в той или иной ситуации».

И все же, чтобы принять диагноз Саши, родителям понадобилось больше года. Евгений Глаголев признается : «Ты оказываешься в ситуации, когда вообще не знаешь, что делать». Он уверен, что важно показать — вот, есть такие люди, кто через это прошел и справился, и готов делиться опытом, советом, помощью.
«Хочется сказать другим родителям, что первое ощущение, что у тебя нет опыта и в тебе только страх, потом пройдет. Это неизбежно. Это просто отсутствие информации. Как только она появится, сразу станет легче… Важно не забывать о себе. А родители всегда забывают о себе, они героически ухаживают за ребенком, но заканчивается это одинаково: без себя ты теряешь себя. И быстро. А потом уходит ощущение жизни. Ты в вечном подвиге, но подвиг вечным быть не может».
Родители Саши говорят, что состояние девочки постоянно очень тяжелое, у нее случаются боли неврологического характера, которые не купируются обычными анальгетиками. Иногда приступ удается снять только при помощи морфина. Но врачи часто считают, что детям такие лекарства противопоказаны. И однажды из-за того, что долго не снимался болевой синдром, у девочки произошел регресс состояния, начались приступы эпилепсии.
Но ребенок все равно ребенок, и Глаголевы замечают у дочери много забавного: «Наша Сашенька по характеру — настоящая маленькая женщина и это видно во всем ее поведении, манерах. Она очень смешно закрывает рукой глаза, это выглядит как «я устала, все свободны». Если она чего-нибудь не хочет, вы от нее не добьетесь этого».
Евгений Глаголев не задается вопросом, сколько времени отведено Саше: «Мы думали об этом, но это гадание на кофейной гуще. Если Саша уйдет — значит, наша жизнь будет какой-то другой».
Он написал в своем блоге:
«Мне близка мысль, что у каждого на этой земле своя миссия. И когда она закончена, тогда человек умирает. Неважно, молодой ты или старый: если все что мог/нужно было сделать — сделано, значит «пора-пора». Саша много раз могла умереть. Но сегодня мы проводим нашу девятую осень вместе. В сентябре всей семьей пережили ковид. Когда он нагрянул в 2020 году, внутри пробежало: «Неужели?!». Ведь очевидно, что Саша со своими больными легкими не перенесет его. Но нет. Улыбаюсь в очередной раз. И не могу не думать про только Ее миссию. Значит вот так. И у меня еще есть время, чтобы быть рядом. И любить здесь».
Вам может быть интересно:
Читайте также: